In silico proteome analysis to facilitate proteomics experiments using mass spectrometry (original) (raw)
- Research
- Open access
- Published: 13 August 2003
Proteome Science volume 1, Article number: 5 (2003)Cite this article
- 29k Accesses
- 4 Altmetric
- Metrics details
Abstract
Proteomics experiments typically involve protein or peptide separation steps coupled to the identification of many hundreds to thousands of peptides by mass spectrometry. Development of methodology and instrumentation in this field is proceeding rapidly, and effective software is needed to link the different stages of proteomic analysis. We have developed an application, proteogest, written in Perl that generates descriptive and statistical analyses of the biophysical properties of multiple (e.g. thousands) protein sequences submitted by the user, for instance protein sequences inferred from the complete genome sequence of a model organism. The application also carries out in silico proteolytic digestion of the submitted proteomes, or subsets thereof, and the distribution of biophysical properties of the resulting peptides is presented. proteogest is customizable, the user being able to select many options, for instance the cleavage pattern of the digestion treatment or the presence of modifications to specific amino acid residues. We show how proteogest can be used to compare the proteomes and digested proteome products of model organisms, to examine the added complexity generated by modification of residues, and to facilitate the design of proteomics experiments for optimal representation of component proteins.
Introduction
Proteomics involves the large-scale or global analysis of the protein complement of an organism [1–3]. The convergence of several factors has led to the rapid emergence of proteomics as a distinct and promising scientific field, notably the completion of genome sequencing projects and advances in sensitive high-throughput protein analysis methods such as mass spectrometry (MS). Proteomics studies can generate massive amounts of experimental data. A single bacterial cell may produce 4000 proteins whose abundances and activities may vary throughout an experiment, while the number of proteins expressed in higher eukaryotes is likely to be at least 10-fold greater. Attempts to catalogue, visualize, and analyze proteomics experiments have therefore become a major challenge. In fact, the development of practical software applications suitable for theoretical and experimental analysis of the proteome lags far behind that for the analysis of genomes and DNA.
A fundamental operation of proteomics is to identify proteins. For most high-throughput applications, proteins are cleaved with site-specific reagents, for example cyanogen bromide (CNBr) or proteases (usually trypsin), to generate smaller peptides better suited to analysis by MS. In shotgun proteomics studies, entire mixtures of proteins are digested. Most proteomics experiments involve four steps: a) protein isolation from a biological sample (e.g. a cell extract) following some experimental treatment; b) fractionation of the resulting proteins (or peptides, the products of proteome digestion) by methods such as two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) or liquid chromatography (LC); c) protein or peptide detection by MS; d) protein identification through manual interpretation or database correlation of mass spectra. Integration of these steps is essential for a successful proteome experiment yet relies on accurate knowledge of the parameters influencing each step. Tools that effectively link the predicted proteome or digested proteome to the data obtained in proteomics experiments are therefore necessary for several reasons. First, they provide a statistical framework that can facilitate interpretation of the output of protein identification algorithms such as MS-Fit and MS-Tag [4] or SEQUEST [5]. Second, failure to observe an expected protein in a proteomics experiment may be for several reasons, including limits to MS detection technology, poor expression or recovery of the protein, or because the protein identification algorithm is inefficient. While recent studies have confirmed the presence of many proteins previously only predicted by their cognate DNA sequences [6], likewise a large number of predicted protein species have never been observed, and software tools can highlight experimental factors that might contribute to these discrepancies. Third, while the genomic DNA sequence is believed to contain all the information needed to describe the protein products of the cell, our knowledge of non-canonical proteins (i.e. proteins other than those that are defined by uninterrupted start and stop codons and whose component residues are unmodified) is very incomplete. The set of canonical proteins for a given organism will probably be expanded several-fold by the phenomena of post-transcriptional splicing and post-translational modification. Software that can analyze data on a whole proteome scale is required for examining such expanded proteomes.
Programs that can analyze several aspects of protein biochemistry and structure are available at websites such as the Swiss Institute of Bioinformatics http://www.isb-sib.ch and the European Bioinformatics Institute http://www.ebi.ac.uk/proteome/. These programs are generally not suited to processing of whole proteomes, nor are they designed to analyze the peptide digestion products of entire proteomes. A software application that can mimic proteome digestion and analyze the resulting peptides on a whole proteome scale would be of great value to the mass spectrometry researcher. We therefore developed proteogest, a program that generates basic descriptive statistics for both the intact and proteolytically processed proteome.
Results
An analytical tool for proteomics
proteogest is written in Perl and runs in command line mode with several options. A detailed description of how to install and run proteogest is available for download at http://www.utoronto.ca/emililab/program/proteogest.htm
Protein sequences to be analyzed are saved as a text file in FASTA format in the same directory as the proteogest program. Text files can be edited to suit the user, for instance to contain all the proteins predicted for a particular organism, or a similar list with predicted transmembrane proteins removed. The user specifies the cleavage criteria by inserting an 'X' character into the cleavage sequence e.g. "SXS" would cleave in the middle of two successive S residues. Where alternative residues may be cleaved, the alternatives are separated by a comma, "PX,QX,RX". The 'Z' character can be used as a wild card, for instance "QZZYZQXS" would mimic the tobacco etch virus protease recognition site where cleavage occurs after the second glutamine (Q) and tolerates several different residues at positions 2, 3, and 5. In the laboratory, the activity of proteolytic enzymes and chemical reagents may be incomplete, resulting in a subset of digestion products that contain cleavage sites that remain unprocessed. In order to simulate this, an option to specify the maximum number of missed cleavages per digestion product is included. When this option is chosen, the output describes all possible complete and incomplete cleavages. For instance, by choosing "2", all peptides containing 0, 1 or 2 missed cleavages are described (not just those where 2 cleavage sites are present).
Several post-translational modification options may be used. A peptide can be modified by phosphorylation (in this case, +80.0 amu can be added to every occurrence of serine, threonine or tyrosine, or iteratively to only one of each separate STY residues) or the user can specify any combination of custom modifications. A number of groups have described promising methods for phosphoproteome analysis recently [7–9] (reviewed in reference 10).
Modifications using quantitative chemical adducting reagents such as the ICAT (Isotope-Coded Affinity Tag; [11, 12]) or MCAT (Mass-Coded Abundance Tag; [13]) are also available and the program can be run in modes that assume all (considers all specified residues as modified) or some (considers specified residues occurring in both modified and unmodified form) proteins or proteins digestion products as modified. This mimics the natural proteome environment where proteins may exist in differentially modified states. The user may vary the output file format. 'Simple' reports (Fig. 1A) and 'Annotated' reports (Fig. 1B) are text files that contain descriptive lists of the proteome digestion products, and can serve as the input for other programs, for a database, or for computational or visualization applications. These reports can be very large (for instance, the set of 25,931 known or predicted human proteins produces 1,175,015 peptides when digested with trypsin) so they are not produced by default. The 'Summary' report is an html document that provides an overview of the processed proteome using descriptive statistics of the parent proteins and the digested daughter peptides. The mean, standard deviation, and range of properties such as protein length, molecular mass are noted and the distribution of these physical properties and properties such as amino acid occurrence are described in a series of tables (Fig. 1C). In another section, similar properties are described for the proteome digestion products (Fig. 1D). These data are useful for researchers planning proteomics experiments using many different fractionation and detection approaches. For example, an investigator planning a HPLC-MS experiment following peptide capture with a tryptophan-binding reagent needs to know both the frequency of tryptophan-containing peptides among parent proteins and the size distribution of those peptides, the former to estimate the representation of the captured peptides and the latter to focus the efforts of the mass spectrometer to the correct mass-to-charge (m/z) range. As well as length, mass, and amino acid representation, charge is predicted for each peptide. The report ends with descriptions of potential phosphorylation and user-defined modifications, as well as statistics describing the extent of redundancy (the occurrence of the same peptide sequence two or more times) among the products.
Figure 1
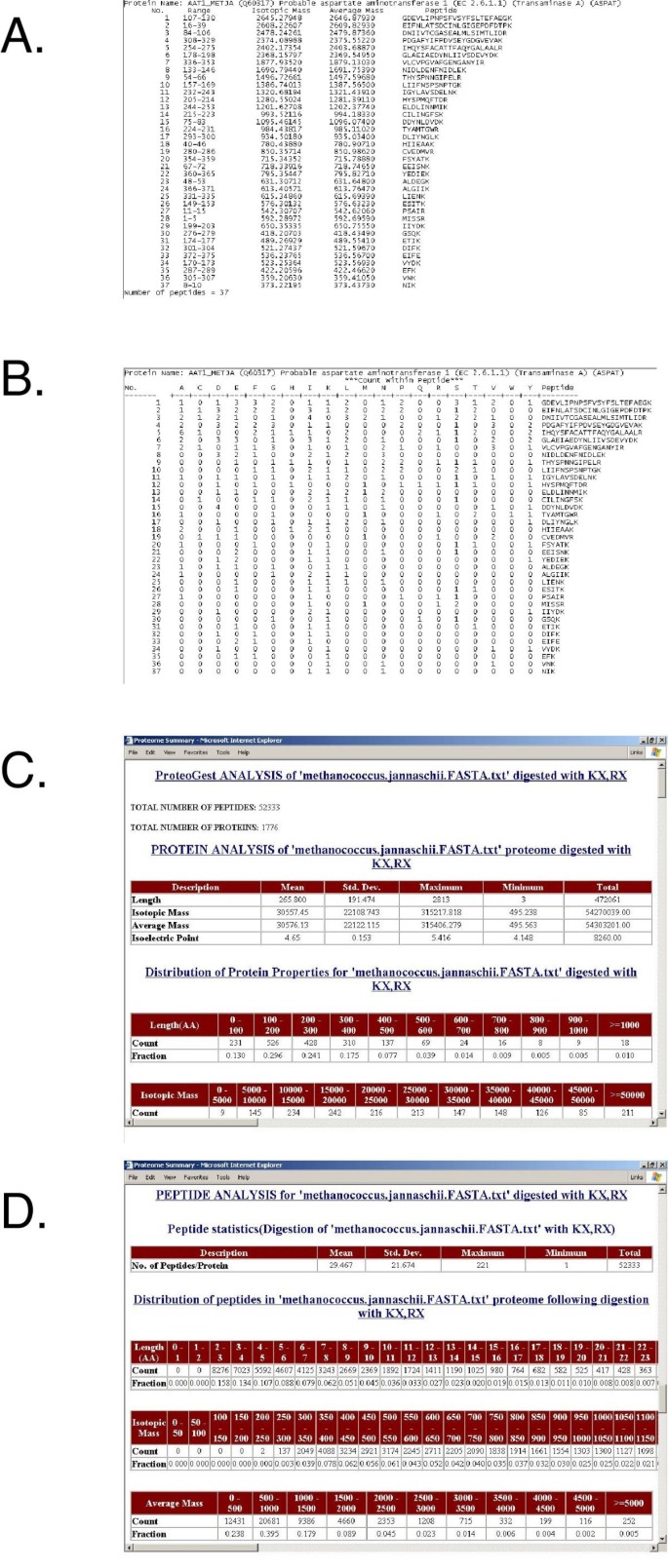
Screen captures of the proteogest output files. The Methanococcus jannaschii proteome was digested C-terminal to lysine and arginine residues (cleavage criteria "KX,RX"). A) The 'Simple' file lists the peptide products of each protein, their isotopic and average molecular masses, and their sequence; B) The 'Annotated' file also lists the peptide products of each protein, along with the counts of each amino acid residue for each peptide; C) A screen capture from the Protein Analysis section of the 'Summary' file; D) A screen capture from the Peptide Analysis section of the 'Summary' file.
Computational proteome analysis of model organisms
proteogest was written to analyze large proteome amino acid sequence datasets and to simulate digestion of the proteome with enzymes or chemical reagents. Here we refer to the theoretical proteome as the entire potential protein complement encoded by the genetic component of a cell or organism, and distinguish it from the observed or experimental proteome, or the complement of proteins that are actually expressed under physiological or experimental conditions. This definition of the proteome includes the primary gene products defined by start and stop codons but does not exclude variants of those gene products arising from mRNA splicing or post-translational proteolysis or modification.
We used proteogest to compare the distribution of protein characteristics for the proteomes of a number of model organisms, including both eukaryotes and prokaryotes. Although the distribution of protein size (M_r_) is roughly similar for all seven organisms examined, eukaryotic organisms tend to express larger proteins than prokaryotes, with for example the average human protein having mass 51,801 Da while a typical E. coli protein is 35,005 Da (Fig. 2A). Next, we compared the occurrence of the 20 amino acids for each protein in the complete predicted proteome sets. For clarity, analyses of only three proteomes, E. coli, S. cerevisiae and H. sapiens, are shown (Fig. 2B). The columns for each amino acid residue are arranged in the figure from left to right according to their frequency in yeast, and it is observed that while some amino acids are favoured (e.g. leucine) or disfavoured (e.g. tryptophan) in all three organisms, there is considerable variation in occurrence. Because the most common method of digesting proteins into peptides in proteomics experiments is the use of trypsin, it is interesting to note that the combined frequency of the amino acids lysine and arginine (trypsin generally cleaves carboxy-terminal to lysine and arginine) varies from a low of 8.8% of all amino acids in E. coli up to 12.1% in M. jannaschii (data not shown).
Figure 2
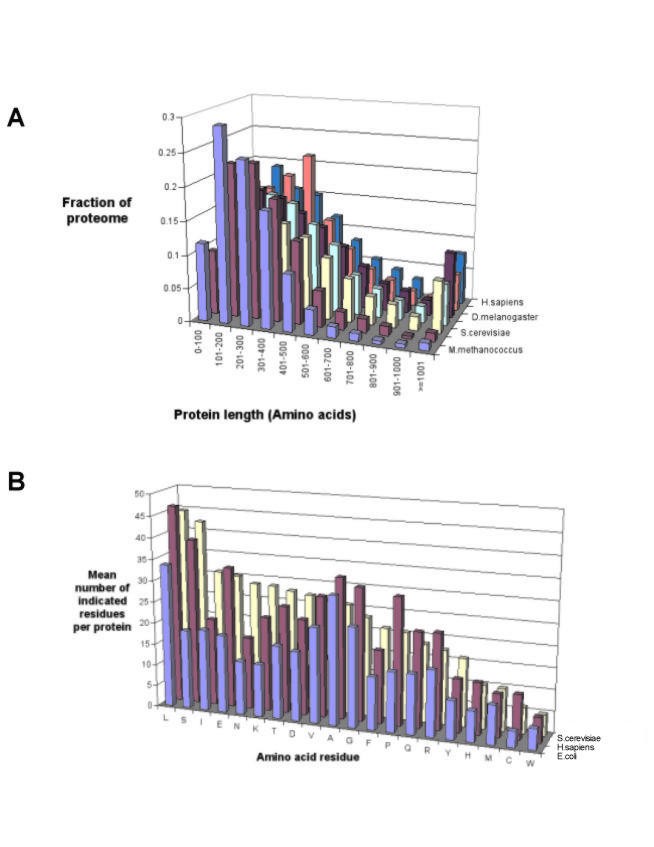
Characteristics of proteins in the predicted proteomes of model organisms. A) Distribution of protein length; B) Mean amino acid residue frequency per protein.
For many laboratory proteomics procedures, it is useful to know exactly how specific residues are distributed. Figure 3 shows that the distribution of different amino acids among proteins varies significantly in the model organisms. The distribution of a common residue like alanine is clearly different from that of a relatively rare residue like tryptophan in the proteomes of the organisms examined. Where the distribution appears to differ between organisms, for instance the distribution of serine, they can generally be accounted for by differences in average protein size. For instance, the fraction of proteins having less than 20 serine residues per protein is almost double for E. coli than for S. cerevisiae or H. sapiens (the average number of serines per protein in these organisms is 18.25, 42.22 and 38.81 respectively), but the corresponding protein lengths are 305.58, 458.27 and 460.41.
Figure 3
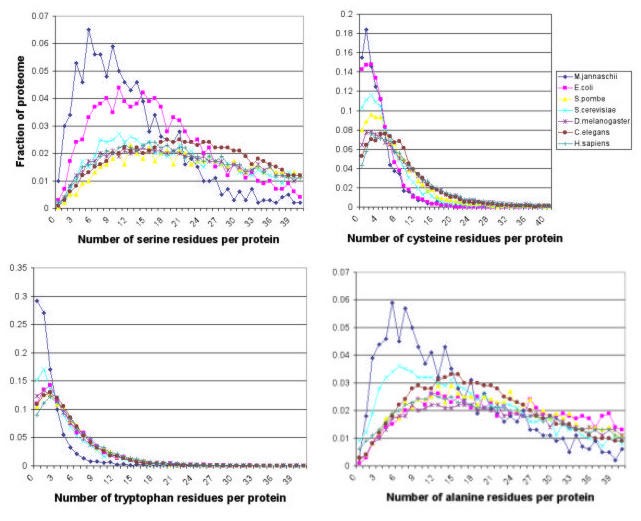
Distribution of alanine-, tryptophan-, serine-, and cysteine-containing proteins in predicted proteomes of model organisms.
Another use of proteogest is for searching proteome datasets for potential binding sites and consensus sequences. For instance, metal affinity capture using Nickel conjugated resins is often used to recover recombinant proteins from E. coli and S. cerevisiae. Interestingly, the sequence HHHHHH (His6) occurs only once in the predicted E. coli proteome (the His operon attenuator leader peptide) while it is present in 17 predicted S. cerevisiae proteins. In contrast, only two E. coli proteins (multidrug resistance protein B and hypothetical protein yciQ) and four S. cerevisiae proteins (AFG3, SCJ1, YLR338W and hypothetical protein YJE8) contain polyglutamine tracts longer than five residues.
The experimentally determined proteome is likely to reflect biases introduced by the methodology used to characterize it. To assess this, we used proteogest to compare the predicted S. cerevisiae proteome to FASTA files representing three sets of proteomic data obtained using the approaches most commonly used in published proteomics studies. One set contains 157 proteins identified following separation on 2D gels [14, 15] and another contains 164 proteins identified in our laboratory using microflow liquid chromatography coupled online to tandem mass spectrometry. A third set contains 1436 proteins that were identified in multi-dimensional protein identification technology (MudPIT) experiments ([6], G. Cagney & A. Emili; unpublished). In MudPIT, successive LC online fractionation steps are carried out on fractions eluted from strong cation exchange media packed in the same chromatography column that houses reverse phase media [16]. MudPIT therefore represents an orthogonal 2D separation technology and is capable of separating many hundreds to thousands of peptides in a single experiment. Comparison of the sets shows clear biases in the methodologies (Fig. 4). Small proteins (<200 amino acids) were poorly recovered from 2D gels, whereas the size profile of proteins identified following 1D reverse phase chromatography more closely resembled that of the proteome (all predicted yeast proteins). Proteins with high isoelectric point were also poorly represented in proteome samples fractionated using 2D gels. The MudPIT approach was perhaps most representative of the proteome, with a similar profile for proteins above 300 residues in length, although significantly fewer proteins in the 100–200 amino acid range are seen using MudPIT than are expected from the predicted proteome. This may result from larger proteins giving proportionally more positive identifications because there are more peptides per protein for the mass spectrometer to detect. This effect is not as noticeable for 2D gel or 1D chromatography because the numbers of proteins normally detected per experiment (~150) is significantly lower than for MudPIT experiments (~1500). These conclusions are supported by protein abundance estimates (Fig. 4C). Of the proteome analysis methods examined, the distribution of codon adaptation indices (CAI) for proteins detected by MudPIT most closely resembled the distribution predicted from the theoretical proteome, although only about half the proportion of very poorly expressed proteins (CAI < 0.15) are observed in MudPIT samples compared to the theoretical proteome. Separation of proteome components using 2D gels also appears capable of identifying moderately abundant proteins, while 1D LC appears to be mostly limited to abundant proteins. The absence of a second fractionation dimension and the relatively short analysis time explain the underperformance of 1D LC when analyzing highly complex proteomic mixtures; these levels of sensitivity and ease of use may be perfectly adequate when analyzing less complex mixtures however. This type of analysis is possible using proteogest, where sets of proteins from any source can be analyzed for simple biophysical properties like digestion product size distribution.
Figure 4
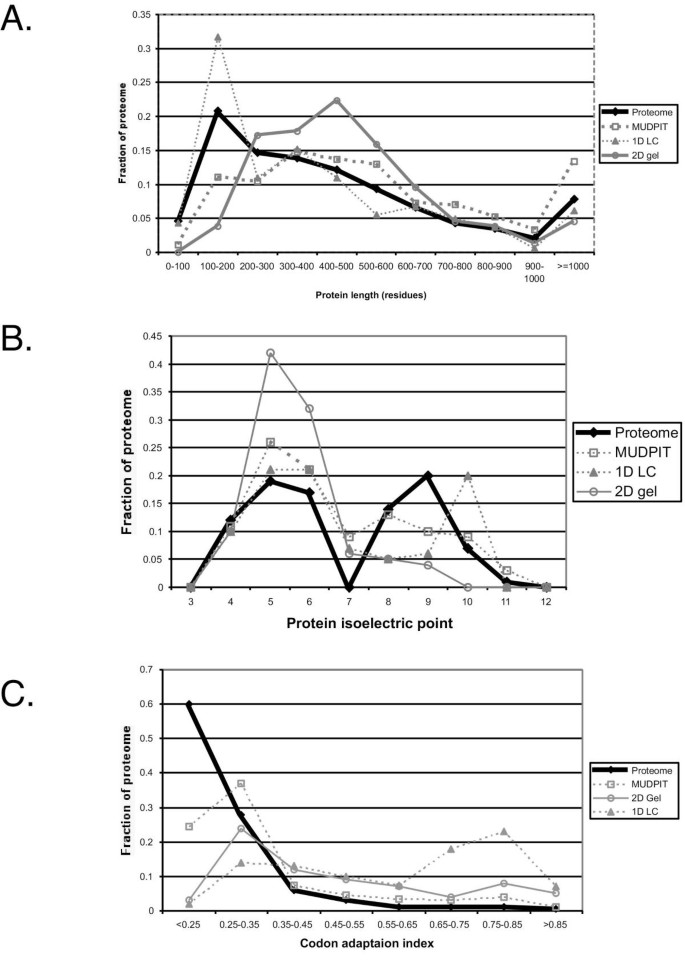
Comparison of methods for identifying experimental proteomes. Experimental proteomes were determined using MALDI MS or nano-electrospray MS following 2D gel protein separation (2D gel), 1D liquid chromatography peptide separation with nano-electrospray MS (1D LC), or 2D peptide separation by 2D chromatography (strong cation exchange and reverse phase chromatography) with nano-electrospray MS (MUDPIT). 'Proteome' represents all predicted yeast proteins. Protein length in residues (A), isoelectric point (B), and codon adaption index, CAI (C), are shown. MUDPIT data were obtained from reference 6 and our laboratory; 1D LC data are from our laboratory; 2D gel data are from references 14 and 15.
Analysis of the digested and modified proteome
We next used proteogest to compare the digestion products of the proteome of S. cerevisiae using reagents commonly used to generate peptides. Treatment of the proteome with trypsin (cleaves C-terminal to lysine or arginine, except before proline), chymotrypsin (cleaves C-terminal to hydrophobic residues), Endo Asp N (cleaves N-terminal to aspartic acid), or cyanogen bromide (cleaves C-terminal to methionine when the reaction is carried out in formic acid) result in different product properties (Table 1). On average, treatment of with chymotrypsin yields an average of 35.0 peptides per protein compared with 9.6 peptides per protein following treatment with cyanogen bromide, reflecting the frequency of the corresponding cleavage sites in the yeast proteome. The mean product mass following digestion with trypsin (41.9 peptides per protein for S. cerevisiae) is just over 1000 Da for all organisms tested (data not shown), a convenient size for analysis by commercial time-of-flight and ion trap mass spectrometers. Combination digestions are sometimes used in the lab, for instance cleavage by cyanogen bromide followed by trypsin – these combinations can also be modelled by proteogest. Note however, that when one missed trypsin cleavage per product was permitted in a simulated digestion of the yeast proteome using proteogest, the mean peptide size increased to 1637.4 Da while the complexity of the mixture (total number of distinct peptides) doubled. Permitting two missed trypsin cleavages continues this trend, yielding mean peptide size of 2078.5 and tripling the total number of peptides compared to the completely digested proteome. Missed cleavages more closely resemble the situation in the laboratory, where it is difficult to ensure complete trypsin digestion of complex mixtures.
Table 1 Characteristics of peptide products following digestion of the yeast proteome with different agents.
The distribution of proteome digestion product masses is of interest to the proteomics investigator because either the ability of the mass analyzer to resolve peptides with similar mass-to-charge (m/z) ratio, or the instrument time needed to sample all the peptides, or both, are often saturated. The experimenter must therefore focus instrument resources on the regions yielding most information. Trypsin is the enzyme of choice for generating peptides for mass spectrometry because it produces peptides with masses compatible with the detection abilities of commonly used mass spectrometers (up to approximately 2000 m/z), while many peptides are long enough to generate useful sequence data by collision induced dissociation. Peptides larger than 2000 Da can be analyzed using ion trap and quadrupole mass analyzers if the peptides have charge greater than one, resulting in m/z ratios within the effective detection range of the instruments. A large number of information poor peptides of less than 700 Da are typically formed by trypsin digestion. When proteogest was used to catalogue the set of predicted tryptic peptides from S. cerevisiae (omitting peptides with six or fewer residues), it was observed that the majority of peptides have mass between 800 and 1200 Daltons (Fig. 5A). In direct contrast, peptides identified in our lab (from several MUDPIT analyses of yeast whole cell protein) are evenly distributed across the range 700–1600 Da (Fig. 5A). It is clear that even strategies broadly representative of the proteome like MudPIT may be saturated at the level of MS detection. This is especially the case for the region 800–1100 Da, where nearly all unit peptide masses are represented at least 30 times. This means that a mass analyzer with unit resolution or less (e.g. many quadrupole and ion trap instruments) cannot resolve these peptides from each other, but even an analyzer capable of resolving to 0.1 Da in the range 800–1100 m/z would face many peptides of similar mass. Certain protein or peptide enrichment protocols may simplify the peptide distributions sufficiently that proteins can be unambiguously determined from one or more daughter peptides. We anticipate that proteogest may be useful in this regard. For instance, relatively rare amino acids (e.g. cysteine, tryptophan) may be used in affinity capture experiments to 'normalize' the proteome because most proteins contain small numbers of these amino acids. However, the proportion of proteins containing no cysteines varies considerably for different proteomes. The human proteome contains just over 4% of proteins lacking cysteine while the corresponding number for M. jannaschii is 15% (Fig. 3). These proteins would be excluded from proteome analysis when using such reagents. We examined the distribution of cysteine- and tryptophan-containing peptides in the trypsin-digested human proteome in more detail (Fig. 5B). While 57.8% of peptides contain at least two cysteines, 93.4% contain at least one (Fig. 5B). The corresponding figures for tryptophan are 35.2% and 89.2% (Fig. 5B). Having more than one peptide per protein means that even if one affinity-trapped peptide remains undetected by the mass spectrometer, a second opportunity for detection may present itself. These figures are useful because they represent a theoretical upper limit to the number of peptides that may be detected by mass spectrometry using these approaches.
Figure 5
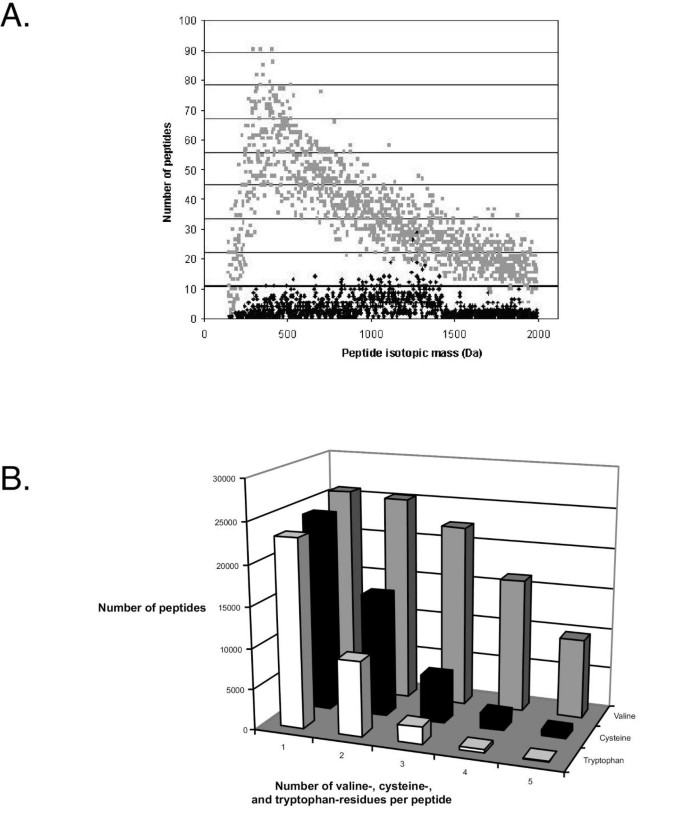
Size distribution of peptides following digestion of the yeast and human proteomes with trypsin. A) The distribution of peptides produced by the theoretical proteome (grey) is compared to that produced in experiment (black). Peptide masses are plotted at unit resolution (Da). The relatively lower numbers of peptides observed between 1600–2000 Da is an artefact resulting from MS runs being programmed to detect only ions with m/z in the range 400–1600. Identification of peptides with mass greater than 1600 is possible with ions of charge greater than one. B) Distribution of valine-, cysteine-, and tryptophan-containing peptides in the human proteome.
The experimentally determined proteome may contain proteins that are naturally modified or that are modified as part of the proteomics experiment. Examples of the former class include acetylation, phosphorylation, and glycosylation while examples of the latter include carboxymethylation and modification by quantitation reagents such as ICAT or MCAT. It is intuitive that the complexity of a mixture of digested proteins will be greatly increased by differential modification, for instance phosphorylation (Fig. 6A). It is currently impossible to accurately predict the effects of chemical modification on the behaviour of the corresponding ions in a mass spectrometer. Of interest to the proteomics researcher however, is to understand how the increased diversity affects the distribution of predictable physical/chemical properties of the digest products. Even with current state-of-the-art 2D chromatography-MS systems, the number of peptides that need to be separated in a proteomics experiment vastly exceeds the resolving ability of the separating chromatography columns and the time available for the mass spectrometer to obtain a spectrum [17, 18]. Calculations using proteogest show how these modifications alter the mass profiles of the modified peptides (Fig. 6B). Two general comments may be made. First, where modification reactions continue to completion, the mass distribution is not significantly altered. The greatest change in mass distribution is in the lower mass ranges, and the change will be proportional to the ratio of the donor group mass to the accepting peptide mass. Addition of a phosphate group to a serine on a peptide of mass 300 increases it's mass by 27%. The region 800–1200 Da that is critical for proteomics MS applications appears to be minimally affected for the treatments simulated by proteogest (Fig. 6B). Note that other effects such as changes in peptide ionization efficiency cannot be simulated may have a major impact on the success of a proteomics experiment. Moreover, when incomplete modification is simulated, the more likely situation under laboratory conditions, the total number of peptides to be analyzed greatly increases (Fig. 6B).
Figure 6
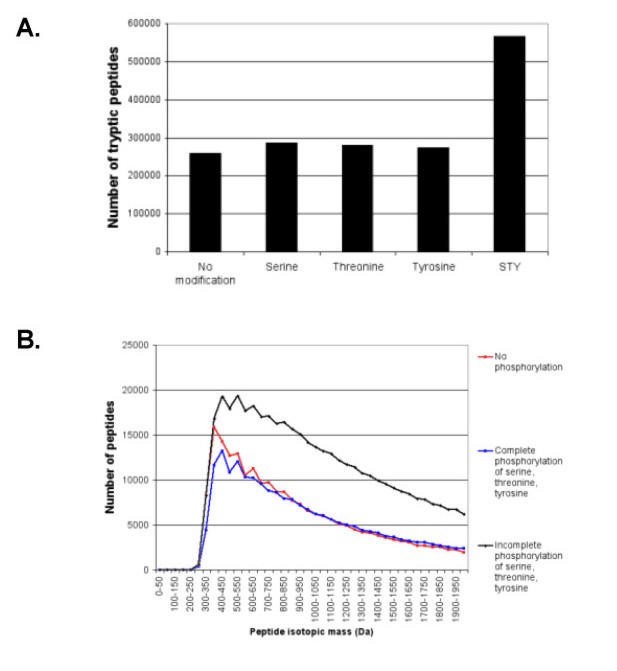
Increased S. cerevisiae proteome complexity resulting from protein modification and incomplete protein cleavage. A) Number of tryptic peptides generated when serine, S, threonine, T, tyrosine, Y, or all three residues are phosphorylated. B) Effect of phosphorylation of serine, threonine, and tyrosine on distribution of tryptic peptide mass.
Many proteins show homology across their whole length or across portions of their sequence, therefore many of the digestion products of such proteins may be identical. The proteogest Redundancy function tabulates redundant peptides at the bottom of the Summary report, listing their sequence along with the cognate protein. Analysis of the E. coli, S. pombe, S. cerevisiae proteomes show distinct differences in the distribution of redundant tryptic peptides (Fig. 7). On average, for every 184 E. coli tryptic peptides (of at least seven amino acids) there is an identical peptide elsewhere in the digested proteome. Meanwhile, for every 49 S. cerevisiae or every 66 S. pombe tryptic peptides, one has an identical counterpart in the proteome. The figure for both yeast species is slightly skewed by the presence of many transposon-derived elements in the genome that are predicted to encode identical proteins. In most cases, common peptides are from protein isoforms, for instance enzyme dimers or the subunits of transport complexes. 898 S. cerevisiae (14.5%), 540 S. pombe (10.7%), and 176 E. coli (4.0%) proteins contain tryptic peptides (> 7 aa) found elsewhere in the proteome, reflecting relative numbers of homologous proteins in these organisms, and perhaps in turn reflecting genome duplication events [19]. From the perspective of the proteomics researcher, it is not possible for peptide sequencing MS approaches to correctly resolve isoforms (or to resolve homologs unless a peptide from one homolog that is not found in a second homolog is observed).
Figure 7
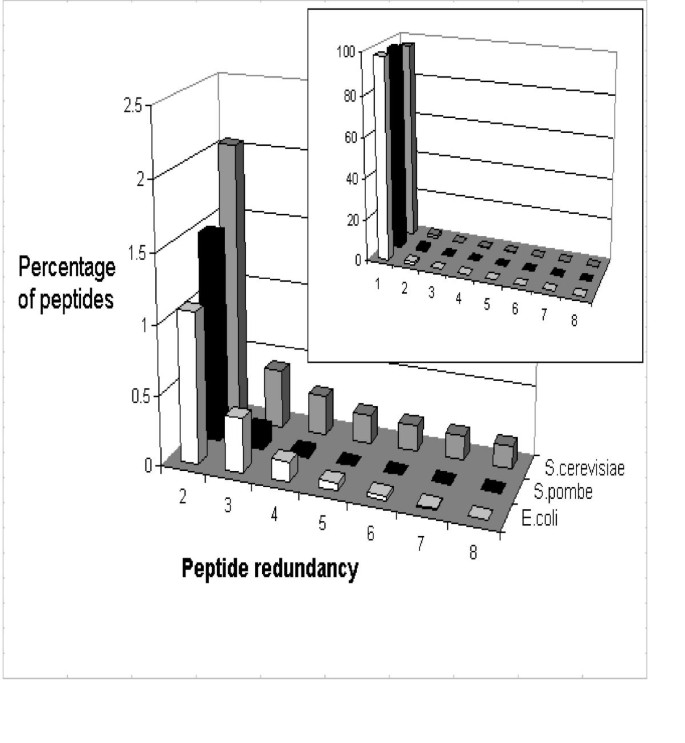
Peptide redundancy in the E. coli, S. pombe, and S. cerevisiae proteomes following in silico digestion with trypsin. Only peptides of seven or more residues are considered.
Finally, protein identifications in proteomics experiments usually rely on measurement of ionized peptides in a mass spectrometer. Different peptides are known to have different ionization properties in particular instruments, yet the chemical or physical basis of this phenomenon is poorly understood [20]. We used proteogest to compare the frequency of different amino acid residues in the predicted tryptic products of the human and mouse proteomes and in greater than 6,000 peptides detected by electrospray mass spectrometry analysis of mouse liver and lung tissue and validated using the SEQUEST and STATQUEST algorithms [21] (Fig. 8). For most residues the differences are minor. Assuming that SEQUEST has no residue bias in matching tandem MS spectra to their cognate peptides, arginine-, serine-, lysine-, and glutamic acid-containing peptides are underrepresented in the identified set, while glycine and particularly histidine are overrepresented. Arginine- and lysine-containing peptides are underrepresented because in silico cleavage forces all such peptides to contain two of either residue (C- and N-terminal), while mass spectrometry experiments often detect peptides that contain only one such flanking residue. Three times as many histidine-containing peptides were identified than would be expected given their frequency in the theoretical proteome. We speculate the histidine-containing peptides are more readily detected during electrospray mass spectrometry because the basic histidine residue promotes ionization (as do lysine and arginine). The rules governing ionization are based on a combination of known and unknown physical and chemical factors and are likely to be subtle, probably requiring examination of the positions as well as frequency of a particular residue in the peptide, or the frequency of different combinations of residues. We believe that proteogest may prove to be useful in addressing this important issue.
Figure 8
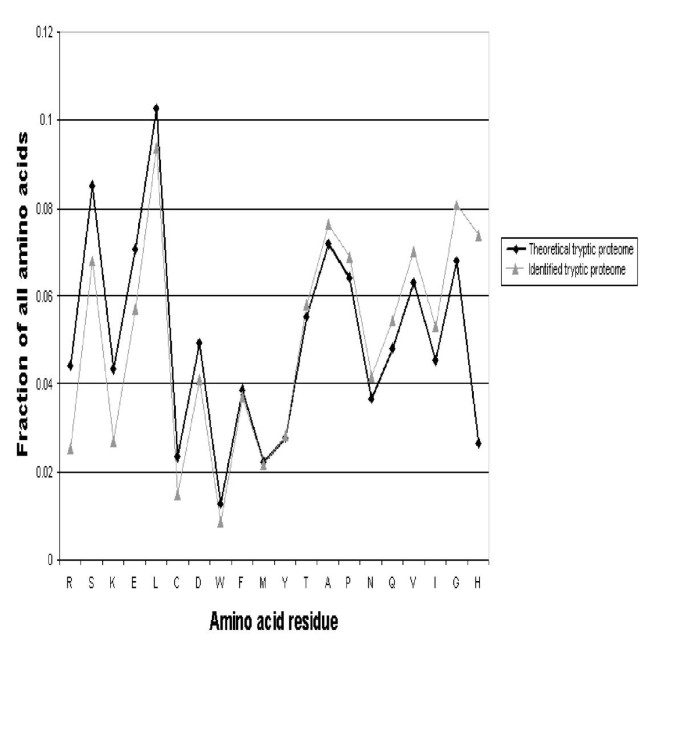
Comparison of amino acid residue frequency in tryptic peptides from lung tissue proteome identified by electrospray mass spectrometry ('Identified tryptic proteome') and in the in silico trypsin digested combined human and mouse proteomes ('Theoretical tryptic proteome'). Residue frequencies are expressed as a fraction of the analyzed proteome (e.g. serine, S, comprises 8.5% of the theoretical tryptic proteome versus 6.8% of the identified tryptic proteome). Note that the occurrence of arginine (R) and lysine (K) are artificially reduced in the Identified tryptic proteome because non fully-tryptic peptides (i.e. peptides with residues other than R or K at their C-termini) form a significant fraction of peptides identified by SEQUEST.
Discussion
Currently, two main MS approaches are used to identify proteins in proteomics experiments: a) 2D-PAGE separation combined with matrix assisted laser desorption ionization MS [22] and b) gel or chromatographic separation combined with electrospray MS [23]. The former approach uses the observed masses of intact peptide ions derived from the same parent protein for identification ("peptide fingerprinting"), while the latter generally relies on uninterpreted product mass spectra derived from a single peptide ion. In both cases, database searching is normally used to match experimentally observed mass spectra with spectra predicted for known protein sequences. The efficiency of both approaches is dependant on many factors, for example the accuracy, sensitivity and resolution of the measuring instrument, and also the size and distribution of peptide and protein properties in proteome. proteogest permits descriptive statistics to be obtained for whole proteome datasets and for in silico digestion products of the proteomes. Normally, calculating these numbers requires a custom program to be written for each query. Although such programs are relatively simple, they require time and skills not always available in a busy proteomics lab. We therefore wrote proteogest to answer questions about the physical/chemical properties of theoretical proteomes, in order to design practical experiments.
The software tool is timely and valuable for several reasons. First, it permits the testing of hypotheses concerning the entire proteome (or large subsets thereof). For instance, one might ask whether yeast nuclear proteins are enriched in particular (e.g. acidic) amino acids by comparing the fraction of certain residues (e.g. aspartic acid and glutamic acid) found in nuclear localized proteins as compared with the overall proteome. To do this, proteogest is first run on a FASTA file of the complete yeast proteome and then on a similar file edited to include only proteins annotated to the nucleus. Second, the distribution of proteins or peptides can be incorporated into probability-based mass spectrum identification algorithms. For instance, the mean number of tryptic peptides per protein for E. coli is 28, but 42 for C. elegans, so the fragmentation patterns expected for a typical protein will the different in the different organisms. Furthermore, automated de novo peptide sequencing (identification of a peptide sequence solely from the spectrum itself, and not by comparison with a spectrum predicted using a DNA database) is currently achievable only using specialized high-resolution mass spectrometers (e.g. Fourier Transform MS) or by chemical modification of the peptides before MS analysis, such as using MCAT [13]. Knowing the relative occurrence of different amino acids (or pairs of successive amino acids) for a given proteome for instance, can facilitate the probability of de novo sequencing predictions. Third, proteogest can be used for the planning and interpretation of experimental proteomics applications, in particular those involving high throughput protein identifications using MS. For instance, the ICAT method for protein relative abundance determination [16] relies on the modification of cysteine-containing peptides. When designing a proteomics experiment using ICAT, it is important to calculate the proportion of all proteins that contain one or more cysteines, yet currently, there is no easy way to carry out this apparently trivial calculation without writing a program. Finally, we show how proteogest can be used to search for patterns in proteomics data, for instance the frequency of particular amino acid residues in observed versus predicted peptides.
Materials and Methods
Datasets
Files containing the protein sequence of all proteins predicted using the genomic DNA sequences of Escherchia coli, Methanococcus jannaschii, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Caenorhabditis elegans, Drosophilia melanogaster, and Homo sapiens were downloaded from the European Bioinformatics website http://www.ebi.org on 20 August 2002. The experimental S. cerevisiae proteome determined by 2D-MALDI was obtained by combining datasets observed by Futcher and coworkers [15] and Gygi and coworkers [14]. The proteome dataset determined by 1D LCMS was obtained from our laboratory using methods described in Cagney and Emili [13]. The 2D MUDPIT proteome dataset comprised proteins observed by Washburn and coworkers [6] and in our laboratory. The experimentally determined sets are subsets of the complete predicted S. cerevisiae proteome FASTA file and proteogest is used in exactly the same way except that the input files are edited to include only relevant proteins. Peptides detected by mass spectrometry following trypsin digestion of whole cell extract of S. cerevisiae were obtained in our laboratory using MUDPIT [16, 21] and identified using the SEQUEST algorithm [5] searched against all predicted fully tryptic peptides in the non-redundant SwissProt and TrEMBL mouse and human protein sequences downloaded from EBI in December 2002. SEQUEST scores demonstrated to yield approximately 98% correct identifications were included in the analysis [21].
proteogest
The software is open source and can be requested by email. The program is written in Perl and works on major operating systems (Windows, Unix, Linux). A helpfile can be downloaded from the Emili website and gives instructions on installing and using the program.
References
- Aebersold R, Mann M: Mass spectrometry-based proteomics. Nature 2003, 422: 198–207. 10.1038/nature01511
Article CAS PubMed Google Scholar - Phizicky E, Bastiaens PIH, Zhu H, Snyder M, Fields S: Protein analysis on a proteomic scale. Nature 2003, 422: 208–215. 10.1038/nature01512
Article CAS PubMed Google Scholar - Tyers M, Mann M: From genomics to proteomics. Nature 2003, 422: 193–197. 10.1038/nature01510
Article CAS PubMed Google Scholar - Clauser KR, Baker PR, Burlingame AL: Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem 1999, 14: 2871–2882. 10.1021/ac9810516
Article Google Scholar - Eng JK, McCormack AL, Yates JR 3rd: An approach to corelate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 1994, 5: 976–989. 10.1016/1044-0305(94)80016-2
Article CAS PubMed Google Scholar - Washburn MP, Wolters D, Yates JR 3rd: Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nature Biotechnol 2001, 19: 242–247. 10.1038/85686
Article CAS Google Scholar - Salomon AR, Ficarro SB, Brill LM, Brinker A, Phung QT, Ericson C, Sauer K, Brock A, Horn DM, Schultz PG, Peters EC: Profiling of tyrosine phosphorylation pathways in human cells using mass spectrometry. Proc Natl Acad Sci USA 2003, 100: 443–448. 10.1073/pnas.2436191100
Article CAS PubMed Central PubMed Google Scholar - Ficarro SB, McCleland ML, Stukenbery PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM: Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nature Biotechnol 2002, 20: 301–305. 10.1038/nbt0302-301
Article CAS Google Scholar - MacCoss MJ, McDonald WH, Saraf A, Sadygov R, Clark JM, Tasto JJ, Gould KL, Wolters D, Washburn M, Weiss A, Clark JI, Yates JR III: Shotgun identification of protein modifications from protein complexes and lens tissue. Proc Natl Acad Sci USA 2002, 99: 7900–7905. 10.1073/pnas.122231399
Article CAS PubMed Central PubMed Google Scholar - Mann M, Jensen ON: Proteomic analysis of post-translational modifications. Nature Biotechnol 2003, 21: 255–261. 10.1038/nbt0303-255
Article CAS Google Scholar - Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R: Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nature Biotechnol 1999, 17: 994–999. 10.1038/13690
Article CAS Google Scholar - Zhou H, Ranish JA, Watts JD, Aebersold R: Quantitative proteome analysis by solid-phase isotope tagging and mass spectrometry. Nature Biotechnol 2002, 19: 512–515. 10.1038/nbt0502-512
Article Google Scholar - Cagney G, Emili A: Do novo peptide sequencing and quantitative profiling of complex protein mixtures using mass-coded abundance tagging. Nature Biotechnol 2002, 20: 163–170. 10.1038/nbt0202-163
Article CAS Google Scholar - Gygi SP, Rochon Y, Franza BR, Aebersold R: Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 1999, 19: 1720–1730.
CAS PubMed Central PubMed Google Scholar - Futcher B, Latter GI, Monardo P, McLaughlin CS, Garrels JI: A sampling of the yeast proteome. Mol Cell Biol 1999, 19: 7357–7368.
CAS PubMed Central PubMed Google Scholar - Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvick BM, Yates JR 3rd: Direct analysis of protein complexes using mass spectrometry. Nature Biotechnol 1999, 17: 676–682. 10.1038/10890
Article CAS Google Scholar - Aebersold R, Goodlett DR: Mass spectrometry in proteomics. Chem Rev 2001, 101: 269–295. 10.1021/cr990076h
Article CAS PubMed Google Scholar - Peng J, Gygi SP: Proteomics: the move to mixtures. J Mass Spectrom 2001, 36: 1083–1091. 10.1002/jms.229
Article CAS PubMed Google Scholar - Wolfe KH, Shields DC: Molecular evidence for an ancient duplication of the entire yeast genome. Nature 1997, 387: 708–713. 10.1038/42711
Article CAS PubMed Google Scholar - Kinter M, Sherman NE: Protein sequencing and identification using tandem mass spectrometry. Wiley-Interscience, New York 1 Edition 2000.
Google Scholar - Kislinger T, Rahman K, Radulovic D, Cox B, Rossant J, Emili A: PRISM, a Generic Large Scale Proteomic Investigation Strategy for Mammals. Mol Cell Proteomics 2003, 2: 96–106. 10.1074/mcp.M200074-MCP200
Article CAS PubMed Google Scholar - Mann M, Hendrickson RC, Pandey A: Analysis of proteins and proteomes by mass spectrometry. Annu Rev Biochem 2001, 70: 437–473. 10.1146/annurev.biochem.70.1.437
Article CAS PubMed Google Scholar - Wu CC, MacCoss MJ: Shotgun proteomics: tools for the analysis of complex biological systems. Curr Opin Mol Ther 2002, 4: 242–250.
CAS PubMed Google Scholar
Acknowledgements
We thank Jimmy Eng, Dave Tabb, and John Yates, III, for generous use of Sequest and DTASelect/Contrast software. We also wish to thank Pete St. Onge, Faye Baron, Duy Mai, and Shaun Ghanny for computing assistance and fruitful suggestions. This work was supported in part by a grant to A.E from the National Science and Engineering Research Council of Canada and Genome Canada.
Author information
Authors and Affiliations
- Program in Proteomics and Bioinformatics, Banting and Best Department of Medical Research, University of Toronto, Toronto, Canada
Gerard Cagney, Shiva Amiri, Thanuja Premawaradena, Micheal Lindo & Andrew Emili - Department of Molecular and Medical Genetics, University of Toronto, Toronto, Canada
Andrew Emili - Department of Clinical Pharmacology, Royal College of Surgeons, 123 Saint Stephen's Green, Dublin 2, Ireland
Gerard Cagney
Authors
- Gerard Cagney
You can also search for this author inPubMed Google Scholar - Shiva Amiri
You can also search for this author inPubMed Google Scholar - Thanuja Premawaradena
You can also search for this author inPubMed Google Scholar - Micheal Lindo
You can also search for this author inPubMed Google Scholar - Andrew Emili
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toAndrew Emili.
Authors’ original submitted files for images
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article: Verbatim copying and redistribution of this article are permitted in all media for any purpose, provided this notice is preserved along with the article's original URL.
About this article
Cite this article
Cagney, G., Amiri, S., Premawaradena, T. et al. In silico proteome analysis to facilitate proteomics experiments using mass spectrometry.Proteome Sci 1, 5 (2003). https://doi.org/10.1186/1477-5956-1-5
- Received: 23 April 2003
- Accepted: 13 August 2003
- Published: 13 August 2003
- DOI: https://doi.org/10.1186/1477-5956-1-5