New Insights of an Old Defense System: Structure, Function, and Clinical Relevance of the Complement System (original) (raw)
- Invited Review Article
- Open access
- Published: 29 October 2010
Molecular Medicine volume 17, pages 317–329 (2011)Cite this article
- 2989 Accesses
- 6 Altmetric
- Metrics details
Abstract
The complement system was discovered a century ago as a potent defense cascade of innate immunity. After its first description, continuous experimental and clinical research was performed, and three canonical pathways of activation were established. Upon activation by traumatic or surgical tissue damage, complement reveals beneficial functions of pathogen and danger defense by sensing and clearing injured cells. However, the latest research efforts have provided a more distinct insight into the complement system and its clinical subsequences. Complement has been shown to play a significant role in the pathogenesis of various inflammatory processes such as sepsis, multiorgan dysfunction, ischemia/reperfusion, cardiovascular diseases and many others. The three well-known activation pathways of the complement system have been challenged by newer findings that demonstrate direct production of central complement effectors (for example, C5a) by serine proteases of the coagulation cascade. In particular, thrombin is capable of producing C5a, which not only plays a decisive role on pathogens and infected/damaged tissues, but also acts systemically. In the case of uncontrolled complement activation, “friendly fire” is generated, resulting in the destruction of healthy host tissue. Therefore, the traditional research that focuses on a mainly positive-acting cascade has now shifted to the negative effects and how tissue damage originated by the activation of the complement can be contained. In a translational approach including structure-function relations of this ancient defense system, this review provides new insights of complement-mediated clinical relevant diseases and the development of complement modulation strategies and current research aspects.
History of the Complement System
The complement system was first recognized in the late 19th century when leading microbiologists such as Paul Ehrlich, Jules Bordet and George Nuttall discovered a bactericidal function of blood on anthrax bacilli (1–4). They noted that this bactericidal function was inactivated when blood was heated up to 55°C or kept at room temperature and named it “alexin.”
Research on guinea pigs demonstrated that the bactericidal activity of blood not only depended on the already described heat-labile alexin, but also on a heat-stable bactericidal factor. In 1899, Paul Ehrlich renamed alexin as complement and called the heat-stable substance amboceptor (3).
By 1920, four components of complement (C1, C2, C3 and C4) had already been detected, each factor being assigned a number in the order in which it had been discovered. Although the order of their discovery did not represent their activation sequence, the names were kept to avoid confusion. The antibody-dependent pathway of complement activation was named the “classical pathway.” Although it had already been discovered in 1913 that some bacteria and yeast as well as cobra venom factor could induce the complement system independently of antibodies, it was not until 1954 that Pillemer discovered the “properdin pathway.” Now known as the “alternative pathway,” it is able to induce the complement cascade independently of antibody interaction by binding directly to bacteria and yeast (5).
Two decades ago, the mannose-binding lectin (MBL), or “lectin activation pathway,” was discovered. Kawasaki et al. (6) found the MBL protein in 1978, but its function remained unclear until 1989, when Super et al. (7) recognized that reduced serum levels of MBL correlated with an opsonic defect in children. Matsushita et al. then detected the proteolytic activity of the MBL-associated serine proteases (MASP-1 and MASP-2), leading to the formation of the classical C3 convertase (8–11).
Paths of Activation and Effects
Established Pathways
Complement activation can occur through three major amplification pathways.
The classical pathway. The classical pathway is antibody-dependent and occurs when circulating antibodies bind to specific pathogens. Only IgM and IgG are capable of sufficient complement activation. After binding of the pathogen, a rearrangement of the crystallizable fragment (Fc)-conformation enables C1q to bind onto the Fc-region of the antibody. Because of the pentamer structure of IgM, one molecule is sufficient to activate the complement. IgG has a monomer structure, and therefore two molecules are required. Binding of C1q activates C1r and leads to cleavage of C1s. Activated C1s can then cleave C4 into the anaphylatoxins C4a and C4b, the latter binding to the surface of the pathogen and activating C2 by splitting it into C2b and C2a. C2b diffuses while C2a remains bound to C4b and together they form the C3 convertase C4b2a.
This convertase now splits C3 into C3a and C3b. C3a then acts as an anaphyla-toxin and diffuses. C3b connects to the C3 convertase and forms the C5 conver-tase C4b2a3b. As well as completion of the C5 convertase, C3b also opsonizes pathogens and therefore promotes phagocytosis. Assembly of the C5 convertase initiates the last phase of the complement cascade, which is identical for all three pathways.
In addition to the antibody-induced activation of the classical pathway, there is also the possibility of antibody-independent activation. It was shown that danger signals such as C-reactive protein, viral proteins, β-amyloid, polyanions (bacterial lipopolysaccharides, DNA and RNA) and mitochondrial fragments, necrotic/apoptotic cells and amyloid P were able to induce the classical pathway (12–15).
The alternative pathway. In contrast to the classical pathway, activation of the alternative pathway proceeds through antibody-independent binding of danger signals such as bacteria, yeast and virus-infected cells, but also protein A, C-reactive protein, cobra venom factor, polysaccharides and damaged tissue (14,16). Because constant activation of the alternative pathway is due to spontaneous hydrolysis of the highly reactive C3, constant control by complement regulators is required (17). Healthy cells are capable of various control mechanisms that prevent the spontaneous activation of C3 and protect the host from undesirable complement activation. These control mechanisms exist both in the fluid phase and membrane bound (see below).
The spontaneous hydrolysis of C3 produces C3(H2O), which functionally resembles C3b. C3(H2O) associates reversibly with factor B, while plasmatic protease factor D cleaves factor B. This event, as well as the small fragment Ba, produces the C3 convertase of the alternative pathway C3(H2O)Bb. Binding of the protein properdin stabilizes the fragment, extending the half-life 10-fold. The C3 convertase then splits C3 into C3a and C3b, with C3b being capable of creating a new C3 convertase with the aid of factor B and D. This amplification loop is highly important not only for the alternative pathway but also for the two other activation pathways.
Binding of more C3b to the C3 convertase now creates the C5 convertase C3(H2O)BbP3b. This initiates the terminal enzymatic cascade of the lytic membrane attack complex (13–15,18).
The lectin pathway. The lectin activation pathway has been rather less intensely studied. Activation takes place when MBL binds mannose-containing surface proteins on pathogenic surfaces. In its ultra-structure, MBL closely resembles C1q, and along with the serine proteases MASP-1 and -2 (which themselves resemble C1r and C1s, respectively), forms a potent multi-enzyme complex (Figure 1).
Figure 1
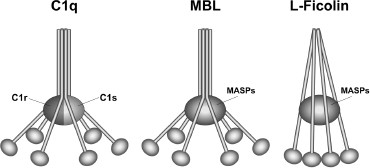
Scheme of the similar structure of complement activation molecules C1q of the classical pathway, MBL of the lectin pathway and L-ficolin.
Upon activation, MASP-2 catalyzes the cleavage of C2 and C4 in a similar manner to the classical pathway and forms a C3 convertase named C4b2a. MASP-1 is capable of C2 and C3 cleavage, although to a much lesser extent (11,19). Subsequently, C3 is cleaved into C3a and C3b, and by accretion of C3b to the C3 convertase, the C5 convertase is formed. A third serine protease MASP-3 has a distinct function compared with MASP-1 and -2, exerting inhibitory actions against MASP-2 (20).
In addition to the established activation of the lectin pathway via MBL and MASPs, it was demonstrated that ficolins were also capable of initiating the lectin pathway by forming active complexes with MASPs. There are three distinct ficolins named ficolin-1 (M-ficolin), ficolin-2 (L-ficolin) and ficolin-3 (H-ficolin or Hakata antigen). Structurally homologous to the collectin MBL as well as C1q, ficolins are soluble collagen-like proteins that bind to sugar structures presented on microorganisms and dying host cells and consequently activate the innate immune system (8,10,21–23).
Surfactant protein A and D (SP-A, SPD), such as MBL, belong to the collectin family (24), but unlike MBL, they are not able to activate the complement directly.
The impact of the MBL pathway remains to be completely elucidated. It is suspected that its major role takes place during early childhood and in particular during the translational period from the passive immunity provided by the mother’s antibodies to the development of the body’s own mature immunity (25).
The lytic membrane attack complex. The final stage of all three activation pathways is the formation of the lytic membrane attack complex (MAC). In contrast to the three different upstream paths forming a C5 convertase, only the cleavage of C5 into the anaphylatoxin C5a and the active C5b represents an enzymatic step, while the rest of the cascade is solely an accretion of stable proteins. In detail, C5b remains bound to the target cell followed by association of C6, resulting in a hydrophilic complex. By accretion of C7, a conformational change occurs—facilitating a stable linkage by exposure of lipophilic groups. Attachment of C8 with its binding component, C8b, induces the penetration of C8a-g into the lipid double layer of the target cell membrane. The final step toward formation of a stable transmembrane pore with a diameter of 10 angstrom is the binding of 10–15 C9 proteins, which generate a cylindrical structure (Figure 2). Assembly of such a pore may lead to osmotic imbalance through the constant flow of ions, small molecules and water along their concentration gradient, resulting in the lysis of the target cell (26).
Figure 2
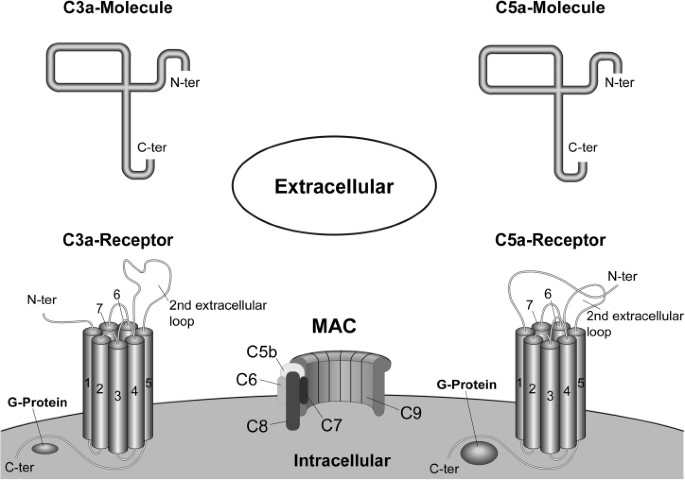
Scheme of MAC with its subdivisions and the two seven-transmembrane-spanning G-protein-coupled receptors C3aR and C5aR. The corresponding ligand (C3a/C5a) is displayed above each receptor.
It is noteworthy that the importance of these transmembrane pores should not be overrated, since, for instance, the blockage of the MAC only leads to a small increase in bacterial Neisseria infection (25,27). The upstream effects of the complement system, such as the anaphylactoid reaction and the opsonization, appear to play a more important role (14,28).
Effects of the complement system. The main effect of the complement system is the induction of a pathogen-associated and modulated enzymatic cascade that, once triggered, ends with the lysis of the target cell and protects the host from infection. In addition to this apparent effect, the complement system also displays crucial additional activities that appear to be even more relevant.
One effect is the opsonization of the pathogen. Cleavage products such as C3b and C4b as well as C5b opsonize the surface of recognized pathogenic substances and therefore facilitate phagocytosis. Additionally, opsonization is also important for the clearance of soluble, circulating antigen-antibody complexes. After the attachment of C3b and C4b to these complexes, they are bound to complement receptor 1 (CR1) on erythrocytes and are subsequently transported to the spleen and liver, where the immune complexes are eliminated.
C3 cleavage products also bridge the innate and the adaptive immune systems. Opsonized antigens are bound to the complement receptor 2 (CR2) on B-cells via the C3-fragment C3d, initiating the production of specific antibodies as well as the differentiation of B-memory cells.
Presumably, the most important function is the induction of an anaphylactoid reaction. The small activation products C3a, C4a and particularly C5a are potent anaphylatoxins, capable of inducing the migration of phagocytes (29), smooth muscle relaxation, degranulation of mast cells and basophile granulocytes and therefore unleashing vasoactive substances such as histamine, prostaglandins, kinins and serotonin. All can cause vasodilation and capillary leakage (30) and induce the migrated cells to release eicosanoids, oxygen radicals and lysosomal enzymes, which cause damage to the pathogens (31–33). It is noteworthy, that complement acts far beyond “inflammation,” as indicated by its close interaction with the coagulation cascade (34–36) and its involvement in the regulation of apoptosis (37–40) and cellular growth (41). The anaphylactoid functions are mediated by the interaction of C3a and C5a with their corresponding seven-transmembrane-spanning receptors C3aR and C5aR (CD88), respectively (see Figure 2). The role of the second C5a receptor named C5L2 is not fully understood and is still controversially discussed. However, there is increasing evidence that C5L2 represents a functional receptor acting as a negative regulator of the inflammatory response. For example, it was shown that inflammation in C5L2 knock-out mice was amplified (42) and that blockage of C5L2 increased serum interleukin (IL)-6 (43).
In summary, complement is highly capable of inducing all classical signs of inflammation, with the occurrence of pain, swelling, reddening, hyperthermia and impaired function.
New Activation Pathways
In addition to the established pathways, new pathways of complement activation were recently discovered (Figure 3).
Figure 3
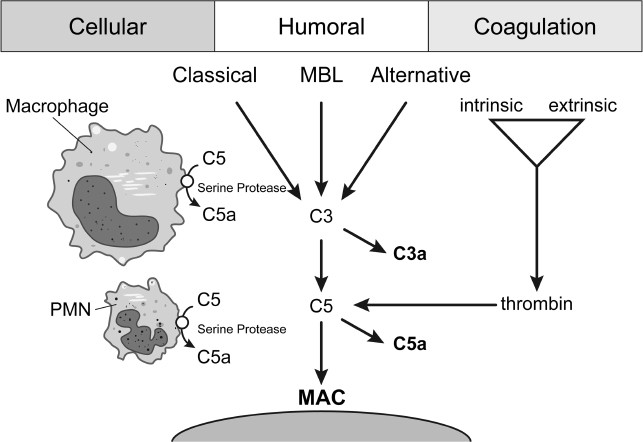
The established pathways of complement activation associated with the new activation pathway and crosstalk between cells and the coagulation cascade with the complement system.
During the last decade, more and more interaction sites between the two major serine protease systems of the human body, namely the coagulation and the complement cascades, were found.
The potent serine protease thrombin is able to directly cleave C3 as well as C5 in a dose- and time-dependent manner, leading to biologically active C3a/C5a (44). In addition to thrombin, investigations by our group indicated proteolytic cleavage of C3 and C5 also by FXa, FXIa and plasmin (35). Interestingly, FVIII and tissue factor failed to directly interact with C3 and C5 (35). Furthermore, FXIIa activates the classical complement pathway via C1q. Crosstalk between the lectin pathway and the coagulation cascade has only recently been ascertained by the observation that the complex of FVIII and von Willebrand factor possesses lectin activity (45).
Vice versa, complement factors also interact with the coagulation system. C1 inhibitor not only blocks all three established complement pathways but also the endogenous coagulation path (46). Ikeda et al. (47) found evidence that C5a induces tissue factor activity on endothelial cells. Furthermore, crosstalk between the anaphylatoxin receptor C5aR and tissue factor was recently found (36,48).
Another possibility for complement activation exists via direct cellular interactions. Huber-Lang et al. demonstrated that phagocytic cells (macrophages, polymorphonuclear leucocytes [PMNs]) are able to cleave C5 into biologically active C5a. This cleavage was conducted by a cell-bound serine protease that was in-ducible for alveolar macrophages, being more constitutively active on PMNs (49).
Regulation of the Complement System
Soluble complement regulators
The complement system can exert manifold detrimental effects not only on pathogenic or damaged tissue but also on healthy host tissue. To protect against a complement attack, the human body has developed various strategies. Principally, there are both membrane-bound and fluid phase complement regulators that are briefly covered (Figure 4).
Figure 4
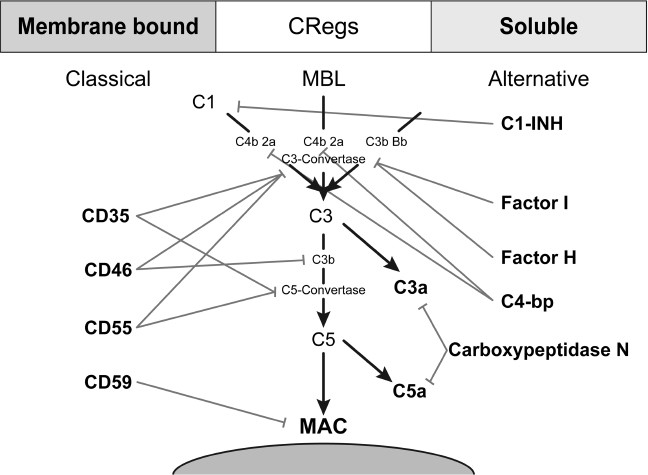
Scheme of the membrane bound and soluble complement regulators acting on different stages of the complement cascade.
The best known regulatory protein is the C1 inhibitor (C1-INH). C1-INH controls the activity of the classical pathway by binding to the C1 complex and initiating the diffusion of the fragments C1r and C1s. This process leads to an irreversible inactivation of the initiating serine protease. As the classical and the lectin pathway resemble each other in many ways, the C1-INH also inactivates MASP-1 and -2, thereby also inhibiting the lectin pathway. As well as inactivating complement components, C1-INH also blocks certain parts of the kinin, fibrinolytic and coagulation systems, such as coagulation factors XII and IX.
Factor I is a serine protease catalyzing the cleavage of the α-chain of C3b and C4b, leading to their permanent inactivation. Cofactors for this enzymatic activity are factor H, C4-binding protein (C4-bp), CD35 and CD46.
Factor H is a protein that hinders the formation of the C3 convertase by competing for the binding site with factor B. Additionally, it facilitates the dissociation of already active C3 convertases and supports the proteolytic cleavage of C3b by factor I.
C4-bp also facilitates the proteolytic cleavage of the α-chain of C4b by factor I in a complex together with protein S.
Serumprotein S (Vitronectin) and clusterin (Sp-40, 40) hinder the formation of the lytic membrane attack complex by adhesion to the lipophilic groups of C7, therefore leading to impaired anchorage in the cell membrane.
Carboxypeptidase N inactivates anaphylatoxins of the complement system as well as other factors such as kinins and creatinine kinase MM through cleavage of terminal arginine and lysine residues of the peptides (17,50).
Membrane-Bound Complement Regulators
CD35 (complement receptor 1 [CR1]) is found on the surface of erythrocytes as well as on leukocytes and on podocytes in the glomerula of the kidney. CD35 facilitates the decay of the C3/C5 convertase and also acts as a cofactor for factor I. CD46 (membrane cofactor protein [MCP]) also acts as a cofactor for factor I-mediated cleavage of C3b and is broadly expressed, except on erythrocytes.
CD55 (decay accelerating factor [DAF]) is widely expressed, except on natural killer cells and on a special subgroup of T-cells. The protein is glycosylphosphatidyl-inositol (GPI) anchored in the cell membrane and accelerates the decay of the classical as well as the alternative C3 convertases by replacing C2a/Bb in these complexes.
CD59 (protectin) is expressed ubiquitously and is similarly integrated into the cell membrane by GPI anchors. It regulates the formation of the terminal lytic membrane attack complex by inhibiting the interaction of the C8α-subchain and the first molecule of C9 so that integration into the cell membrane and the creation of a transmembrane pore is prevented (50–53).
Clinical Relevance and Complement-Mediated Diseases
Sepsis and Complement
In contrast to all the beneficial effects for the host organism, the complement system can also be detrimental for the host tissue (Figure 5).
Figure 5
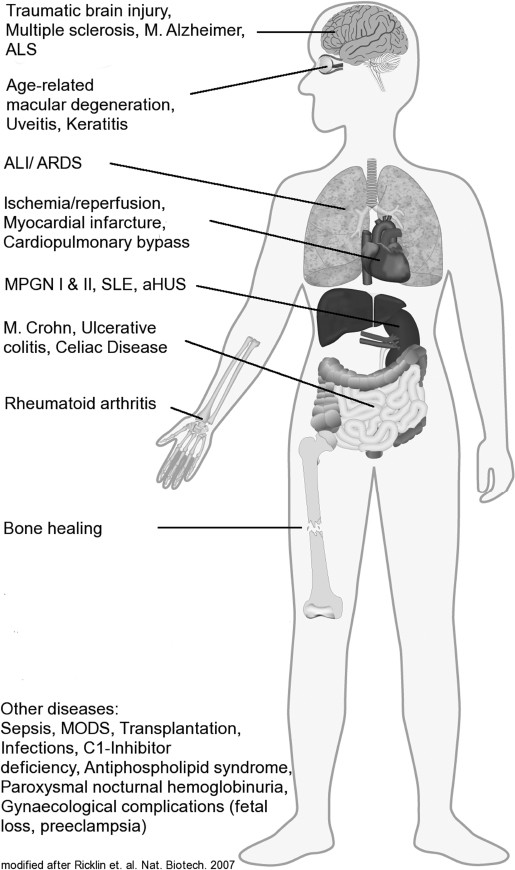
Scheme of diseases in which complement has a major impact. ALS, amyotrophic lateral sclerosis; ALI, acute lung injury; ARDS, adult respiratory distress syndrome; MPGN, membranoproliferative glomerulonephritis; SLE, systemic lupus erythematosus; aHUS, atypical hemolytic uremic syndrome; MODS, multiple organ dysfunction syndrome. Adapted by permission from Macmillan Publishers Ltd: Ricklin D, Lambris JD. (2007) Complement-targeted therapeutics. Image reprinted with permission from Nat. Biotechnol. 25:1265–75.
Many distinct pathogenetic mechanisms may lead to the expression of an excessive and uncontrolled immune response. Depending on the individual’s immune status, this immune response leads to a proinflammatory systemic immune response syndrome or to compensated antiinflammatory response syndrome. Clinical complications of these reactions can be progressive sepsis and the development of multiple organ dysfunction syndrome, with enhanced susceptibility to infections.
Table 1. Different complement-modulating drugs listed by activity level along with references of experimental/clinical application.
Sepsis is defined as a systemic immune response syndrome with signs of infection. Excessive inflammation is induced by the recognition of pathogen-associated molecular patterns on invading microorganisms or danger-associated molecular patterns of damaged tissue by the cellular “first line of defense” and the complement system. This result consequently leads to the robust release of cytokines from phagocytes (“cytokine storm”) to fight the infection. Although of benefit to the organism when acting locally, this pattern leads to a dramatic life-challenging event when occurring systemically (54–58).
Various studies proposed excessive complement activation during sepsis in humans (59). Because of its potent inflammatory profile, C5a appears to be the most detrimental molecule and has been described as “too much of a good thing” (54). When activated, C5a may lead to immune paralysis, multiorgan dysfunction and thymocyte apoptosis (37,40) as well as disturbance of the coagulation and fibrinolytic cascades (34).
In accordance, some protection of septic mice has been shown by the application of a C5a receptor antagonist (60). Furthermore, C5aR antagonism during sepsis led to a changed cytokine profile, such as decreased levels of tumor necrosis factor-α and IL-6, suggesting a direct or indirect role in the synthesis of these factors (40). Czermak et al. (61) showed an enhanced survival rate of septic rats treated with a C5a antibody. Additionally, these authors found that C5a binds to neutrophils, which led to inactivation of their functions.
In a study by Flierl et al. (62), the effects of complement on sepsis using C3−/− and C5−/− deficient mice were examined. In the absence of either C3 or C5, a reduced production of proinflammatory mediators was found.
On a cellular level, it has been shown that C5a effectively interacts with cells and modulates their apoptosis rate. Interestingly, the effects on programmed cell death seem to be cell dependent, with a higher rate of apoptosis in thymocytes (37,40) but decreased apoptosis in neutrophils (39,63,64). Overall, the C5a-induced changes point toward an enhanced susceptibility toward infections, as well as to a prolonged presence of neutrophils resulting in an exaggerated inflammatory response and host damage.
Complement and Bone Biology
In the emerging field of osteoimmunology, the role of complement in bone biology in general and fracture healing in particular has started to raise interest. Although some direct interactions between the immune system and bone cells have been found (65,66), few studies demonstrated the presence of complement components in bone cells. In particular, the expression of various complement factors might depend on the cell differentiation state. Murine os-teoblastic cells were shown to produce C3 in response to vitamin D3 (67,68). During osteoblastic differentiation in murine osteoblasts and in human cell lines, the complement components C1q, C4, C1 inhibitor, C3a receptor (C3aR), properdin and the complement factor H were upregulated (69), whereas the expression of the subcomponents C1r and C1s together with factor H was decreased (70). The expression of functionally active C5a receptor (C5aR; CD88) that modulated IL-6 production was described in a human osteoblastic cell line (71,72). Recently, mRNA and protein expression of C3aR and C5aR in human MSC were reported (73).
Immunohistochemical studies clearly indicated that complement was activated during enchondral ossification, possibly for the modulation of apoptosis (74,75). These studies indicated that some interaction of the complement system with bone cells exists. However, the resulting function to date has been minimally investigated. With the exception of our unpublished data and the immunohisto-chemical studies in enchondral ossification, in vivo data on the expression of complement components in bone are lacking, particularly with regard to fracture healing.
Recently, a major role of an excessively stimulated complement system in the posttraumatic inflammatory response was postulated (16,76,77). Prospective studies with polytraumatized patients showed a consumption and massive activation of complement products, positively correlating with the mortality rate (78–80). Fracture healing is delayed in particular when additional severe injuries stress the organism (81). The massive trauma-induced inflammation correlates with consequent organ dysfunction (79) and may possibly be involved in the delayed fracture healing.
Studies with experimental blunt thoracic trauma found that the complement system contributed to the inflammatory reaction, and additionally successful inhibition of various inflammatory mediators occurred upon application of an antibody against C5a (76). Ganter et al. reported an earlier complement activation after major trauma, for which magnitude correlated with the mortality rate (16). Unpublished data from our group underscore the role of complement in fracture healing, reflected by the enhanced C5aR immunostaining of bone sections.
Traumatic Brain Injury
The anaphylatoxins C3a and C5a have been reported to exert both protective and detrimental effects in the central nervous system (82,83). It is noteworthy that many cells of the central nervous system are more susceptible to a complement attack, since they lack important complement regulators such as CD59 (84). Complement products were demonstrated to be dramatically upregulated in models of cerebral ischemia in rats (85), and evidence for deposition of C3d and C9 after experimental cerebral contusion was found (86). In agreement with these experimental findings, systemic complement activation has been shown in stroke patients (87). A negative role for complement has been proposed in traumatic brain injury, since systemic depletion of complement by infusion of the cobra venom factor improved blood flow and neurological function and reduced cerebral edema during experimental intracerebral hemorrhage (88,89). Moreover, C1 deficiency (90) and complement inhibition by infusion of the C1 inhibitor revealed some neuroprotective effects (91,92). In contradiction to these results for traumatic/ischemic injuries, C1q has a strong neuroprotective effect in neurodegenerative diseases (93). Traumatic brain injury induces C5aR upregulation in an experimental model in rats with enhanced C5a serum levels for >1 week. The functional consequences of traumatic brain injury-induced systemic C5a generation have still to be elucidated (94).
Ischemia/Reperfusion Injuries and Cardiovascular Diseases
After ischemia/reperfusion injury, complement is activated via all three established pathways (classical, alternative and MBL) (95–97). Endothelium damaged by hypoxic stress activates complement, leading to elevated vascular permeability, release of anaphylatoxins and the accumulation of neutrophils (98). Experimental studies indicated complement activation after ischemia/reperfusion in several organs, including lung, liver, gut, kidney, myocardium and skeletal muscle (99–105).
Cardiac surgery and cardiopulmonary bypass surgery are known to cause a considerable immunologic impact to the body, resulting in a systemic inflammatory reaction (106,107). Various factors contribute to the extent of the immunological impact. In addition to general surgical trauma, hypothermia and blood loss, the main mechanisms are exposure of blood to foreign surfaces of the cardiopulmonary bypass, the ischemia/reperfusion injury by aortic cross-clamping and splanchnic hypoperfusion leading to mucosal damage and consequently endotoxemia (108).
An important pathogenetic role for complement in ischemia/reperfusion of the myocardium was indicated experimentally, where inhibition of the complement cascade greatly reduced myocardial damage after myocardial infarction (109–111).
In translational studies, C5 inhibitor pexelizumab was capable of reducing mortality in patients undergoing coronary artery bypass surgery (112) but not myocardial infarction (113). Recently, serum levels of C3a and C5a were found to be significantly elevated in patients suffering from stent restenosis after implantation of drug-eluting stents in patients with coronary heart disease (114).
It was recently found that high MBL levels and low plasma levels of sC5b-9 were associated with an increased risk of dysfunctional cardiac performance after myocardial infarction, possibly owing to the increased complement activity during ischemia/reperfusion, which generally triggers an inflammatory response (115).
However, activation of the complement cascade after ischemia/reperfusion injury or exposure to foreign surfaces is not the only cause of harmful effects of the complement system. Several studies alluded to a role for complement in cardiovascular diseases such as atherosclerosis or vasculitis.
Kawasaki disease, a systemic vasculitis in childhood causing coronary artery aneurysmata, appears to be related to MBL deficiency according to genetic family studies by Biezeveld et al. (116). Rugonfalvi-Kiss et al. (117) found significantly lower restenosis rates in females with low MBL levels undergoing thromboendarteryectomy of the carotid artery because of atherosclerotical stenosis. Additionally, patients with MBL deficiency had a greater risk of myocardial infarction than individuals with normal MBL serum levels (118,119). The proposed protective effect of complement in the pathogenesis of atherosclerosis was experimentally underlined by C3−/− mice exhibiting accelerated development of atherosclerosis (120). Clinical analysis of C2-deficient humans revealed a significant increase in cardiovascular diseases (121). In particular, classical pathway involvement was demonstrated by Bhatia et al. (122) when C1q protected against the development of atherosclerosis in combined C1q and LDLr (low-density-lipoprotein receptor) knockout mice.
In agreement with these results, deficiency of complement inhibitors such as CD59 promoted atherogenesis (123). However, it is noteworthy that both downstream complement defects and activation of the terminal pathway were associated with inherited proatherogenic effects and an increased risk of cardiovascular disease (124).
In conclusion, tightly regulated complement activation seems to protect against atherogenesis, possibly through the clearance of apoptotic cells and other debris, whereas inhibition of the terminal pathway by complement regulators appears to hinder proinflammatory effects (123,125). Once an atherogenic plaque has been formed, increasingly leading to ischemia and reperfusion injury, complement is excessively activated and may lead to harmful tissue damage.
Infections
Generally, a major complication of complement insufficiency is an enhanced susceptibility toward infection. Several studies have shown low MBL serum levels (genetically derived) are correlated with enhanced development of systemic immune response syndrome/sepsis in patients admitted to the intensive care unit (126,127). Low MBL serum concentrations were also associated with the development of pneumonia after surgery in colorectal cancer patients (128). Similarly, MASP-2 deficiency caused increased infection rates with Streptococcus pneumoniae (129). Furthermore, there is evidence for a higher rate of chorioamnionitis in patients with low MBL levels and preterm birth (130). Low MBL serum levels appear to be associated with recurrent spontaneous abortion based on in utero infection. This was proposed by Kruse et al. (131), who demonstrated that patients with MBL levels <100 ng/mL had a higher abortion rate. Some urogenital infections such as vulvovaginitis, herpes simplex virus-2 infection, vulvovaginal candidiasis and vestibulitis appear to be linked to low MBL levels in comparison to healthy controls (132–134). Downstream complement deficiencies are typically associated with recurrent invasive infections of Neisseria meningitidis and gonorrhea (135).
In defects further upstream, such as factor D or properdin, N. meningitidis also appear to be involved in most infections (136). Deficiencies of the classical pathway may also cause enhanced infection rates, since invasive infection was the predominant clinical manifestation in patients with C2 deficiency, especially with S. pneumoniae (121).
Immunomodulation of Complement
Many efforts have been undertaken to effectively modulate the activity of complement, thereby trying to ameliorate or completely abolish the symptoms of complement-mediated diseases. It is tempting to speculate that the effective modulation of the broad spectrum of internal diseases (as listed below) may also reflect an effective future target of various complement-dependent surgical diseases.
C1 inhibitor provided protective effects in myocardial cell injury (137), transplantation (138) and various other diseases (139). C1-INH was effective in a randomized, double-blind clinical study in septic intensive care unit patients. Whereas the renal dysfunction was reduced, the mortality rate remained unchanged in both groups (140). The only clinical application for C1 inhibitor so far is for C1 inhibitor deficiency leading to hereditary angioedema (141).
Nafamostat/FUT-175 is an unspecific serine protease inhibitor that, besides complement activation, also blocks abundant serine protease of the coagulation system (142). It is currently used clinically for the treatment of acute pancreatitis and for the prevention of thrombosis in disseminated intravascular coagulation and extracorporal circulation (143,144), underscoring the crosstalk of the complement and coagulation cascades (35).
Because of a lack of specificity and short half-life, other serine protease inhibitors, such as factor D, were not transferred to clinical trials, and further development was stopped (145,146).
sCR1/TP10 is a soluble complement regulator inhibiting both the classical and alternative pathways and therefore is rather promising regarding ischemia/reperfusion injury and various other conditions. The substance revealed some protective effects in male patients undergoing coronary artery bypass grafting but not in females (145,147). Therefore, clinical trials have been stopped (145). Similar substances such as sCR1-sLex/TP20 with an improved structure (148) and Microcept/APT070 (149) are currently under investigation for the treatment of diseases such as acute myocardial infarction, stroke and inflammatory diseases but have not entered clinical evaluation (150).
A hybrid form of the complement regulators DAF and MCP was designed, named complement activity blocker 2 (CAB2), and entered clinical trials under the name MLN-2222 for the treatment of coronary artery bypass grafting (151).
The complement regulator CD55 (DAF) was recently shown to ameliorate the hepatic inflammation in experimental chronic hepatitis C (152) and autoimmune posterior uveitis (153).
The complement regulator CD59 was investigated in the context of cancer research. Neuroblastoma cells are known to escape cell lysis by abundantly expressing CD59. Therefore, a peptide was generated that was capable of suppressing CD59 expression that resulted in complement-mediated killing of the neuroblastoma cells (154). Experimental studies for the treatment of paroxysmal nocturnal hemoglobinuria with the substitution of CD59 have recently been conducted (155), but definitive results are pending.
C4BP as well as factor H have both been experimentally used to successfully abrogate complement activation on artificial surfaces and to inhibit the development of arthritis. However, these substances have not yet been clinically evaluated (156–158).
Because of their specificity, antibodies against various complement factors appear to be a more reliable treatment strategy.
Eculizumab is a monoclonal antibody against C5 investigated for the treatment of paroxysmal nocturnal hemoglobinuria (159) and is currently in a phase I clinical trial for the treatment of systemic lupus erythematosus (160). The first clinical studies in patients suffering from atypical hemolytic uremic syndrome were conducted and were partly promising, with the optimal dose and timing still to be determined (161).
Pexelizumab is also a monoclonal antibody against C5 and has revealed beneficial effects after cardiopulmonary bypass surgery, but not after myocardial infarction in clinical studies (112,113). Various other antibodies are currently in developmental progress. Neutrazumab and TNX-558 are both directed against C5aR. TNX-234 is an antibody against factor D. TA106 blocks factor B from targeting, for example, in age-dependent macular dys-trophia and asthma (162). At least one antibody directed against properdin is currently undergoing testing (145). In experimental studies, this antiproperdin monoclonal antibody was shown to be beneficial during coronary artery bypass grafting, reducing the activation of platelets and neutrophils (163).
Ofatumumab is a monoclonal antibody against CD20 and exerts its effects not by inhibition, like most other complement therapeutics, but by stimulation of complement-dependent cytotoxicity. It is currently in clinical trials for the treatment of rheumatoid arthritis, B-cell chronic lymphocytic leukemia and follicular lymphoma (164,165). A similar approach that has not yet been clinically tested is the efficacy of the related substances HuMax-CD38 for multiple myeloma and HuMax-ZP3 for the treatment of colon, pancreatic and prostate cancer.
PMX-53 is a peptidic C5aR antagonist and has proven to be advantageous in experimental animal studies for neurode-generative diseases, rheumatoid arthritis, ischemia/reperfusion and inflammatory bowel disease (102,166,167). It is currently being evaluated in clinical trials, but a recent study in humans failed to show significant effects on synovial inflammation (168).
Compstatin, a peptidic C3 inhibitor, is considered a promising drug and is currently in clinical trials for the treatment of age-dependent macular dystrophia (169). Since compstatin has proven to be effective in various animal models for other conditions (170), its application does not appear to be limited to age-dependent macular dystrophia.
It is estimated that 30% of the human population present decreased plasma levels of MBL owing to genetic mutations. Therefore, a recombinant human form of MBL as a substitution therapy for MBL-deficient people was developed and found to be safe (171). The substance is currently under clinical investigation for people suffering from multiple myeloma and undergoing high-dose chemotherapy (145,172,173).
ARC1905 is an aptamer-based C5 inhibitor that inhibits the cleavage of C5 into C5a and C5b. It is intended for intravitreal application in age-dependent macular dystrophia and is currently undergoing phase I clinical trials (174). With JPE-1375 and JSM-7717, there now exists even more C5aR antagonists that are in preclinical evaluation for the treatment of inflammatory, renal and ocular diseases.
Conclusion
Although complement research has been in the center of interest for many years, our understanding and insight into the cascade mechanisms and their complex interaction with other protein cascades such as the coagulation cascade is still in its nascent phase. Currently, complement activation is not divisable into three canonical pathways with separate activation patterns. The exact role of complement in many diseases needs to be further clarified because successful complement interventions need to be matched to the individual patient and be as specific as possible.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
- Bordet J. (1895) Les leukocytes et les proprietes actives du serum chez les vaccines [in French]. Ann. Inst. Pasteur. 9:462–506.
Google Scholar - Bordet J. (1898) Sur l’agglutination et la dissolution des globules rouge par le sérum d’animaux injecteies de sang defibiné [in French]. Ann. Inst. Pasteur. 12:688.
Google Scholar - Ehrlich P, Morgenroth J. (1899) Zur Theorie der Lysenwirkung [in German]. Berlin Klin. Wchsr. 36:6.
Google Scholar - Nuttall G. (1888) Experimente über die bacterien-feindliche Einflüsse des tierischen Körpers [in German]. Z. Hyg. Infecionskir. 4:353.
Google Scholar - Pillemer L, et al. (1954) The properdin system and immunity. I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science 120:279–85.
Article CAS PubMed Google Scholar - Kawasaki T, Etoh R, Yamashina I. (1978) Isolation and characterization of a mannan-binding protein from rabbit liver. Biochem. Biophys. Res. Commun. 81:1018–24.
Article CAS PubMed Google Scholar - Super M, Thiel S, Lu J, Levinsky RJ, Turner MW. (1989) Association of low levels of mannan-binding protein with a common defect of opsonisation. Lancet 2:1236–9.
Article CAS PubMed Google Scholar - Matsushita M, Endo Y, Fujita T. (2000) Cutting edge: complement-activating complex of ficolin and mannose-binding lectin-associated serine protease. J. Immunol. 164:2281–4.
Article CAS PubMed Google Scholar - Matsushita M, Fujita T. (1992) Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J. Exp. Med. 176:1497–502.
Article CAS PubMed Google Scholar - Matsushita M, et al. (2002) Activation of the lectin complement pathway by H-ficolin (Hakata antigen). J. Immunol. 168:3502–6.
Article CAS PubMed Google Scholar - Matsushita M, Thiel S, Jensenius JC, Terai I, Fujita T. (2000) Proteolytic activities of two types of mannose-binding lectin-associated serine protease. J. Immunol. 165:2637–42.
Article CAS PubMed Google Scholar - Gewurz H, Ying SC, Jiang H, Lint TF. (1993) Nonimmune activation of the classical complement pathway. Behring Inst. Mitt. 138–47.
- Barrington R, Zhang M, Fischer M, Carroll MC. (2001) The role of complement in inflammation and adaptive immunity. Immunol. Rev. 180:5–15.
Article CAS PubMed Google Scholar - Gasque P. (2004) Complement: a unique innate immune sensor for danger signals. Mol. Immunol. 41:1089–98.
Article CAS PubMed Google Scholar - Thurman JM, Holers VM. (2006) The central role of the alternative complement pathway in human disease. J. Immunol. 176:1305–10.
Article CAS PubMed Google Scholar - Ganter MT, et al. (2007) Role of the alternative pathway in the early complement activation following major trauma. Shock 28:29–34.
Article CAS PubMed Google Scholar - Liszewski MK, Farries TC, Lublin DM, Rooney IA, Atkinson JP. (1996) Control of the complement system. Adv. Immunol. 61:201–83.
Article CAS PubMed Google Scholar - Harboe M, Mollnes TE. (2008) The alternative complement pathway revisited. J. Cell. Mol. Med. 12:1074–84.
Article CAS PubMed PubMed Central Google Scholar - Hajela K, et al. (2002) The biological functions of MBL-associated serine proteases (MASPs). Immunobiology 205:467–75.
Article CAS PubMed Google Scholar - Dahl MR, et al. (2001) MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity 15:127–35.
Article CAS PubMed Google Scholar - Liu Y, et al. (2005) Human M-ficolin is a secretory protein that activates the lectin complement pathway. J. Immunol. 175:3150–6.
Article CAS PubMed Google Scholar - Matsushita M, Endo Y, Hamasaki N, Fujita T. (2001) Activation of the lectin complement pathway by ficolins. Int. Immunopharmacol. 1:359–63.
Article CAS PubMed Google Scholar - Matsushita M, Fujita T. (2001) Ficolins and the lectin complement pathway. Immunol. Rev. 180:78–85.
Article CAS PubMed Google Scholar - Day AJ. (1994) The C-type carbohydrate recognition domain (CRD) superfamily. Biochem. Soc. Trans. 22:83–8.
Article CAS PubMed Google Scholar - Walport MJ. (2001) Complement. First of two parts. N. Engl. J. Med. 344:1058–66.
Article CAS PubMed Google Scholar - Dalmasso AP, Falk RJ, Raij L. (1989) The pathobiology of the terminal complement complexes. Complement Inflamm. 6:36–48.
Article CAS PubMed Google Scholar - Walport MJ. (2001) Complement. Second of two parts. N. Engl. J. Med. 344:1140–4.
Article CAS PubMed Google Scholar - Morgan BP. (1989) Mechanisms of tissue damage by the membrane attack complex of complement. Complement Inflamm. 6:104–11.
Article CAS PubMed Google Scholar - Marder SR, Chenoweth DE, Goldstein IM, Perez HD. (1985) Chemotactic responses of human peripheral blood monocytes to the complement-derived peptides C5a and C5a des Arg. J. Immunol. 134:3325–31.
CAS PubMed Google Scholar - Schumacher WA, Fantone JC, Kunkel SE, Webb RC, Lucchesi BR. (1991) The anaphylatoxins C3a and C5a are vasodilators in the canine coronary vasculature in vitro and in vivo. Agents Actions 34:345–49.
Article CAS PubMed Google Scholar - Goldstein IM, Weissmann G. (1974) Generation of C5-derived lysosomal enzyme-releasing activity (C5a) by lysates of leukocyte lysosomes. J. Immunol. 113:1583–88.
CAS PubMed Google Scholar - Mollnes TE, et al. (2002) Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood 100:1869–77.
CAS PubMed Google Scholar - Sacks T, Moldow CF, Craddock PR, Bowers TK, Jacob HS. (1978) Oxygen radicals mediate endothelial cell damage by complement-stimulated granulocytes. An in vitro model of immune vascular damage. J. Clin. Invest. 61:1161–67.
Article CAS PubMed PubMed Central Google Scholar - Laudes IJ, et al. (2002) Anti-c5a ameliorates coagulation/fibrinolytic protein changes in a rat model of sepsis. Am. J. Pathol. 160:1867–75.
Article CAS PubMed PubMed Central Google Scholar - Amara U, et al. (2008) Interaction between the coagulation and complement system. Adv. Exp. Med. Biol. 632:71–9.
CAS PubMed PubMed Central Google Scholar - Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. (2007) Complement and coagulation: strangers or partners in crime? Trends Immunol. 28:184–92.
Article CAS PubMed Google Scholar - Guo RF, et al. (2000) Protective effects of anti-C5a in sepsis-induced thymocyte apoptosis. J. Clin. Invest. 106:1271–80.
Article CAS PubMed PubMed Central Google Scholar - Markiewski MM, et al. (2009) The regulation of liver cell survival by complement. J. Immunol. 182:5412–18.
Article CAS PubMed Google Scholar - Perianayagam MC, Balakrishnan VS, King AJ, Pereira BJ, Jaber BL. (2002) C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int. 61:456–63.
Article CAS PubMed Google Scholar - Riedemann NC, et al. (2002) C5a receptor and thymocyte apoptosis in sepsis. FASEB J. 16:887–8.
Article CAS PubMed Google Scholar - Markiewski MM, et al. (2008) Modulation of the antitumor immune response by complement. Nat. Immunol. 9:1225–35.
Article CAS PubMed PubMed Central Google Scholar - Gerard NP, et al. (2005) An anti-inflammatory function for the complement anaphylatoxin C5a-binding protein, C5L2. J. Biol. Chem. 280:39677–80.
Article CAS PubMed Google Scholar - Gao H, et al. (2005) Evidence for a functional role of the second C5a receptor C5L2. FASEB J. 19:1003–5.
Article CAS PubMed Google Scholar - Huber-Lang M, et al. (2006) Generation of C5a in the absence of C3: a new complement activation pathway. Nat. Med. 12:682–7.
Article CAS PubMed Google Scholar - Santizo F, et al. (2009) Lectin activity of the coagulation factor VIII/von Willebrand complex. Tohoku J. Exp. Med. 217:209–15.
Article CAS PubMed Google Scholar - Davis AE 3rd. (2004) Biological effects of C1 inhibitor. Drug News Perspect. 17:439–46.
Article CAS PubMed Google Scholar - Ikeda K, et al. (1997) C5a induces tissue factor activity on endothelial cells. Thromb. Haemost. 77:394–8.
Article CAS PubMed Google Scholar - Ritis K, et al. (2006) A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J. Immunol. 177:4794–802.
Article CAS PubMed Google Scholar - Huber-Lang M, et al. (2002) Generation of C5a by phagocytic cells. Am. J. Pathol. 161:1849–59.
Article CAS PubMed PubMed Central Google Scholar - Meri S, Jarva H. (1998) Complement regulation. Vox Sang. 74 Suppl 2:291–302.
Article CAS PubMed Google Scholar - Morgan BP. (1995) Complement regulatory molecules: application to therapy and transplantation. Immunol. Today 16:257–9.
Article CAS PubMed Google Scholar - Miwa T, Song WC. (2001) Membrane complement regulatory proteins: insight from animal studies and relevance to human diseases. Int. Immunopharmacol. 1:445–59.
Article CAS PubMed Google Scholar - Kim DD, Song WC. (2006) Membrane complement regulatory proteins. Clin. Immunol. 118:127–36.
Article CAS PubMed Google Scholar - Gerard C. (2003) Complement C5a in the sepsis syndrome: too much of a good thing? N. Engl. J. Med. 348:167–9.
Article CAS PubMed Google Scholar - Guo RF, Ward PA. (2006) C5a, a therapeutic target in sepsis. Recent Pat. Antiinfect. Drug Discov. 1:57–65.
Article CAS PubMed Google Scholar - Rittirsch D, Flierl MA, Ward PA. (2008) Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 8:776–87.
Article CAS PubMed PubMed Central Google Scholar - Ward PA. (2004) The dark side of C5a in sepsis. Nat. Rev. Immunol. 4:133–42.
Article CAS PubMed Google Scholar - Ward PA. (2008) Sepsis, apoptosis and complement. Biochem. Pharmacol. 76:1383–8.
Article CAS PubMed PubMed Central Google Scholar - Riedemann NC, Guo RF, Ward PA. (2003) Novel strategies for the treatment of sepsis. Nat. Med. 9:517–24.
Article CAS PubMed Google Scholar - Huber-Lang MS, et al. (2002) Protection of innate immunity by C5aR antagonist in septic mice. FASEB J. 16:1567–74.
Article CAS PubMed Google Scholar - Czermak BJ, et al. (1999) Protective effects of C5a blockade in sepsis. Nat. Med. 5:788–92.
Article CAS PubMed Google Scholar - Flierl MA, et al. (2008) Functions of the complement components C3 and C5 during sepsis. FASEB J. 22:3483–90.
Article CAS PubMed PubMed Central Google Scholar - Guo RF, et al. (2006) In vivo regulation of neutrophil apoptosis by C5a during sepsis. J. Leukoc. Biol. 80:1575–83.
Article CAS PubMed Google Scholar - Perianayagam MC, Balakrishnan VS, Pereira BJ, Jaber BL. (2004) C5a delays apoptosis of human neutrophils via an extracellular signal-regulated kinase and Bad-mediated signalling pathway. Eur. J. Clin. Invest. 34:50–6.
Article CAS PubMed Google Scholar - Arron JR, Choi Y. (2000) Bone versus immune system. Nature 408:535–36.
Article CAS PubMed Google Scholar - Takayanagi H. (2007) Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 7:292–304.
Article CAS PubMed Google Scholar - Hong MH, et al. (1991) Transcriptional regulation of the production of the third component of complement (C3) by 1 alpha,25-dihydroxyvitamin D3 in mouse marrow-derived stromal cells (ST2) and primary osteoblastic cells. Endocrinology 129:2774–79.
Article CAS PubMed Google Scholar - Sato T, et al. (1991) The specific production of the third component of complement by osteoblastic cells treated with 1 alpha,25-dihydroxyvitamin D3. FEBS Lett. 285:21–24.
Article CAS PubMed Google Scholar - Roman-Roman S, et al. (2003) Identification of genes regulated during osteoblastic differentiation by genome-wide expression analysis of mouse calvaria primary osteoblasts in vitro. Bone 32:474–82.
Article CAS PubMed Google Scholar - Billiard J, et al. (2003) Transcriptional profiling of human osteoblast differentiation. J. Cell. Biochem. 89:389–400.
Article CAS PubMed Google Scholar - Jiang T, Gao H. (2008) Expression of C5a receptor in osteoblasts [abstract]. FASEB J. 22:1121.12.
Google Scholar - Pobanz JM, Reinhardt RA, Koka S, Sanderson SD. (2000) C5a modulation of interleukin-1 beta-induced interleukin-6 production by human osteoblast-like cells. J. Periodontal Res. 35:137–45.
Article CAS PubMed Google Scholar - Schraufstatter IU, Discipio RG, Zhao M, Khal-doyanidi SK. (2009) C3a and C5a are chemotactic factors for human mesenchymal stem cells, which cause prolonged ERK1/2 phosphorylation. J. Immunol. 182:3827–36.
Article CAS PubMed Google Scholar - Andrades JA, et al. (1996) Complement proteins are present in developing endochondral bone and may mediate cartilage cell death and vascularization. Exp. Cell. Res. 227:208–13.
Article CAS PubMed Google Scholar - Sakiyama H, et al. (1997) Complement Cls, a classical enzyme with novel functions at the endochondral ossification center: immunohistochemical staining of activated Cls with a neoantigen-specific antibody. Cell Tissue Res. 288:557–65.
Article CAS PubMed Google Scholar - Flierl MA, et al. (2008) The role of C5a in the innate immune response after experimental blunt chest trauma. Shock 29:25–31.
CAS PubMed Google Scholar - Galvan MD, et al. (2008) Deficiency in complement C1q improves histological and functional locomotor outcome after spinal cord injury. J. Neurosci. 28:13876–88.
Article CAS PubMed PubMed Central Google Scholar - Donnelly TJ, et al. (1994) Cytokine, complement, and endotoxin profiles associated with the development of the adult respiratory distress syndrome after severe injury. Crit. Care Med. 22:768–76.
Article CAS PubMed Google Scholar - Keel M, Trentz O. (2005) Pathophysiology of polytrauma. Injury 36:691–709.
Article PubMed Google Scholar - Zilow G, Joka T, Obertacke U, Rother U, Kirschfink M. (1992) Generation of anaphylatoxin C3a in plasma and bronchoalveolar lavage fluid in trauma patients at risk for the adult respiratory distress syndrome. Crit. Care Med. 20:468–73.
Article CAS PubMed Google Scholar - Bhandari M, et al. (2003) Predictors of reoperation following operative management of fractures of the tibial shaft. J. Orthop. Trauma 17:353–61.
Article PubMed Google Scholar - van Beek J, et al. (2001) Complement anaphylatoxin C3a is selectively protective against NMDA-induced neuronal cell death. Neuroreport 12:289–93.
Article PubMed Google Scholar - Osaka H, Mukherjee P, Aisen PS, Pasinetti GM. (1999) Complement-derived anaphylatoxin C5a protects against glutamate-mediated neurotoxicity. J. Cell. Biochem. 73:303–11.
Article CAS PubMed Google Scholar - Bradt BM, Kolb WP, Cooper NR. (1998) Complement-dependent proinflammatory properties of the Alzheimer’s disease beta-peptide. J. Exp. Med. 188:431–8.
Article CAS PubMed PubMed Central Google Scholar - Schafer MK, et al. (2000) Complement C1q is dramatically up-regulated in brain microglia in response to transient global cerebral ischemia. J. Immunol. 164:5446–52.
Article CAS PubMed Google Scholar - Pasinetti GM, et al. (1992) Complement C1qB and C4 mRNAs responses to lesioning in rat brain. Exp. Neurol. 118:117–25.
Article CAS PubMed Google Scholar - Pedersen ED, Waje-Andreassen U, Vedeler CA, Aamodt G, Mollnes TE. (2004) Systemic complement activation following human acute ischaemic stroke. Clin. Exp. Immunol. 137:117–22.
Article CAS PubMed PubMed Central Google Scholar - Vasthare US, et al. (1998) Complement depletion improves neurological function in cerebral ischemia. Brain Res. Bull. 45:413–9.
Article CAS PubMed Google Scholar - Xi G, Hua Y, Keep RF, Younger JG, Hoff JT. (2001) Systemic complement depletion diminishes perihematomal brain edema in rats. Stroke 32:162–7.
Article CAS PubMed Google Scholar - Ten VS, et al. (2005) C1q-deficiency is neuroprotective against hypoxic-ischemic brain injury in neonatal mice. Stroke 36:2244–50.
Article CAS PubMed Google Scholar - De Simoni MG, et al. (2004) The powerful neuroprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. Am. J. Pathol. 164:1857–63.
Article PubMed PubMed Central Google Scholar - Longhi L, et al. (2009) C1-inhibitor attenuates neurobehavioral deficits and reduces contusion volume after controlled cortical impact brain injury in mice. Crit. Care Med. 37:659–65.
Article CAS PubMed Google Scholar - Benoit M, Tenner AJ. (2010) Regulation of neuronal gene and miRNA expression by the complement protein C1q associated with neuroprotection. Mol. Immunol. 47:2250–51.
Article Google Scholar - Stahel PF, Kossmann T, Morganti-Kossmann MC, Hans VH, Barnum SR. (1997) Experimental diffuse axonal injury induces enhanced neuronal C5a receptor mRNA expression in rats. Brain Res. Mol. Brain Res. 50:205–12.
Article CAS PubMed Google Scholar - Arumugam TV, Shiels IA, Woodruff TM, Granger DN, Taylor SM. (2004) The role of the complement system in ischemia-reperfusion injury. Shock 21:401–9.
Article CAS PubMed Google Scholar - Link C, et al. (1999) Selection of phage-displayed anti-guinea pig C5 or C5a antibodies and their application in xenotransplantation. Mol. Immunol. 36:1235–47.
Article CAS PubMed Google Scholar - Stahl GL, et al. (2003) Role for the alternative complement pathway in ischemia/reperfusion injury. Am. J. Pathol. 162:449–55.
Article CAS PubMed PubMed Central Google Scholar - Arumugam TV, et al. (2006) Complement mediators in ischemia-reperfusion injury. Clin. Chim. Acta. 374:33–45.
Article CAS PubMed Google Scholar - Buerke M, Murohara T, Lefer AM. (1995) Cardioprotective effects of a C1 esterase inhibitor in myocardial ischemia and reperfusion. Circulation 91:393–402.
Article CAS PubMed Google Scholar - Jaeschke H, Farhood A, Bautista AP, Spolarics Z, Spitzer JJ. (1993) Complement activates Kupffer cells and neutrophils during reperfusion after hepatic ischemia. Am. J. Physiol. 264:G801–9.
CAS PubMed Google Scholar - Proctor LM, et al. (2004) Comparative anti-inflammatory activities of antagonists to C3a and C5a receptors in a rat model of intestinal ischaemia/reperfusion injury. Br. J. Pharmacol. 142:756–64.
Article CAS PubMed PubMed Central Google Scholar - Arumugam TV, et al. (2003) A small molecule C5a receptor antagonist protects kidneys from ischemia/reperfusion injury in rats. Kidney Int. 63:134–42.
Article CAS PubMed Google Scholar - De Vries B, et al. (2003) Inhibition of complement factor C5 protects against renal ischemia-reperfusion injury: inhibition of late apoptosis and inflammation. Transplantation 75:375–82.
Article PubMed Google Scholar - Kyriakides C, et al. (1999) Skeletal muscle reperfusion injury is mediated by neutrophils and the complement membrane attack complex. Am. J. Physiol. 277:C1263–8.
Article CAS PubMed Google Scholar - Kyriakides C, et al. (2000) Neutrophil mediated remote organ injury after lower torso ischemia and reperfusion is selectin and complement dependent. J. Trauma. 48:32–8.
Article CAS PubMed Google Scholar - Ascione R, et al. (2000) Inflammatory response after coronary revascularization with or without cardiopulmonary bypass. Ann. Thorac. Surg. 69:1198–204.
Article CAS PubMed Google Scholar - Butler J, Rocker GM, Westaby S. (1993) Inflammatory response to cardiopulmonary bypass. Ann. Thorac. Surg. 55:552–9.
Article CAS PubMed Google Scholar - Laffey JG, Boylan JF, Cheng DC. (2002) The systemic inflammatory response to cardiac surgery: implications for the anesthesiologist. Anesthesiology 97:215–52.
Article CAS PubMed Google Scholar - Langlois PF, Gawryl MS. (1988) Detection of the terminal complement complex in patient plasma following acute myocardial infarction. Atherosclerosis 70:95–105.
Article CAS PubMed Google Scholar - Mathey D, et al. (1994) Early accumulation of the terminal complement-complex in the ischaemic myocardium after reperfusion. Eur. Heart J. 15:418–23.
Article CAS PubMed Google Scholar - Vakeva A, et al. (1994) Time course of complement activation and inhibitor expression after ischemic injury of rat myocardium. Am. J. Pathol. 144:1357–68.
CAS PubMed PubMed Central Google Scholar - Mahaffey KW, et al. (2006) Effect of pexelizumab on mortality in patients with acute myocardial infarction or undergoing coronary artery bypass surgery: a systematic overview. Am. Heart J. 152:291–6.
Article CAS PubMed Google Scholar - Testa L, et al. (2008) Pexelizumab in ischemic heart disease: a systematic review and meta-analysis on 15,196 patients. J. Thorac. Cardiovasc. Surg. 136:884–93.
Article CAS PubMed Google Scholar - Speidl WS, et al. (2010) Coronary late lumen loss of drug eluting stents is associated with increased serum levels of the complement components C3a and C5a. Atherosclerosis 208:285–9.
Article CAS PubMed Google Scholar - Haahr-Pedersen S, et al. (2009) Level of complement activity predicts cardiac dysfunction after acute myocardial infarction treated with primary percutaneous coronary intervention. J. Invasive Cardiol. 21:13–9.
PubMed Google Scholar - Biezeveld MH, et al. (2003) Association of mannose-binding lectin genotype with cardiovascular abnormalities in Kawasaki disease. Lancet 361:1268–70.
Article CAS PubMed Google Scholar - Rugonfalvi-Kiss S, et al. (2005) High rate of early restenosis after carotid eversion endarterectomy in homozygous carriers of the normal mannose-binding lectin genotype. Stroke 36:944–8.
Article CAS PubMed Google Scholar - Madsen HO, Videm V, Svejgaard A, Svennevig JL, Garred P. (1998) Association of mannose-binding-lectin deficiency with severe atherosclerosis. Lancet 352:959–60.
Article CAS PubMed Google Scholar - Saevarsdottir S, et al. (2005) Mannan binding lectin as an adjunct to risk assessment for myocardial infarction in individuals with enhanced risk. J. Exp. Med. 201:117–25.
Article CAS PubMed PubMed Central Google Scholar - Buono C, et al. (2002) Influence of C3 deficiency on atherosclerosis. Circulation 105:3025–31.
Article CAS PubMed Google Scholar - Jönnson G, et al. (2005) Homozygous C2 deficiency in Sweden: frequent occurrence of invasive infection, atherosclerosis and rheumatic disease. Medicine (Baltimore) 84:23–34.
Article Google Scholar - Bhatia VK, et al. (2007) Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am. J. Pathol. 170:416–26.
Article CAS PubMed PubMed Central Google Scholar - Yun S, Leung VW, Botto M, Boyle JJ, Haskard DO. (2008) Brief report: accelerated atherosclerosis in low-density lipoprotein receptor-deficient mice lacking the membrane-bound complement regulator CD59. Arterioscler. Thromb. Vasc. Biol. 28:1714–6.
Article CAS PubMed Google Scholar - Schmiedt W, et al. (1998) Complement C6 deficiency protects against diet-induced atherosclerosis in rabbits. Arterioscler. Thromb. Vasc. Biol. 18:1790–5.
Article CAS PubMed Google Scholar - Oksjoki R, et al. (2007) Complement regulation in human atherosclerotic coronary lesions: immunohistochemical evidence that C4b-binding protein negatively regulates the classical complement pathway, and that C5b-9 is formed via the alternative complement pathway. Atherosclerosis 192:40–8.
Article CAS PubMed Google Scholar - Fidler KJ, et al. (2004) Increased incidence and severity of the systemic inflammatory response syndrome in patients deficient in mannose-binding lectin. Intensive Care Med. 30:1438–45.
Article PubMed Google Scholar - Garred P, Strom J, Quist L, Taaning E, Madsen HO. (2003) Association of mannose-binding lectin polymorphisms with sepsis and fatal outcome, in patients with systemic inflammatory response syndrome. J. Infect. Dis. 188:1394–403.
Article CAS PubMed Google Scholar - Ytting H, Christensen IJ, Jensenius JC, Thiel S, Nielsen HJ. (2005) Preoperative mannan-binding lectin pathway and prognosis in colorectal cancer. Cancer Immunol. Immunother. 54:265–72.
Article CAS PubMed Google Scholar - Stengaard-Pedersen K, et al. (2003) Inherited deficiency of mannan-binding lectin-associated serine protease 2. N. Engl. J. Med. 349:554–60.
Article CAS PubMed Google Scholar - Annells MF, et al. (2005) Polymorphisms in immunoregulatory genes and the risk of histologic chorioamnionitis in Caucasoid women: a case control study. BMC Pregnancy Childbirth 5:4.
Article PubMed CAS PubMed Central Google Scholar - Kruse C, et al. (2002) Low serum level of mannan-binding lectin is a determinant for pregnancy outcome in women with recurrent spontaneous abortion. Am. J. Obstet. Gynecol. 187:1313–20.
Article CAS PubMed Google Scholar - Babula O, Danielsson I, Sjoberg I, Ledger WJ, Witkin SS. (2004) Altered distribution of mannose-binding lectin alleles at exon I codon 54 in women with vulvar vestibulitis syndrome. Am. J. Obstet. Gynecol. 191:762–6.
Article CAS PubMed Google Scholar - Babula O, Lazdane G, Kroica J, Ledger WJ, Witkin SS. (2003) Relation between recurrent vulvovaginal candidiasis, vaginal concentrations of mannose-binding lectin, and a mannose-binding lectin gene polymorphism in Latvian women. Clin. Infect. Dis. 37:733–7.
Article PubMed Google Scholar - Gadjeva M, et al. (2004) Mannan-binding lectin modulates the response to HSV-2 infection. Clin. Exp. Immunol. 138:304–11.
Article CAS PubMed PubMed Central Google Scholar - Figueroa JE, Densen P. (1991) Infectious diseases associated with complement deficiencies. Clin. Microbiol. Rev. 4:359–95.
Article CAS PubMed PubMed Central Google Scholar - Sjöholm AG. (2002) Deficiencies of mannose-binding lectin, the alternative pathway, and the late complement components. In: Rose NR, Hamilton RG, Detrick B (eds.) Manual of Clinical Laboratory Immunology. ASM Press, Washington, DC, pp. 847–54.
Google Scholar - Fu J, et al. (2006) Anti-ischemia/reperfusion of C1 inhibitor in myocardial cell injury via regulation of local myocardial C3 activity. Biochem. Biophys. Res. Commun. 350:162–8.
Article CAS PubMed Google Scholar - Kirschfink M. (2002) C1-inhibitor and transplantation. Immunobiology 205:534–41.
Article CAS PubMed Google Scholar - Kirschfink M. (2001) Targeting complement in therapy. Immunol. Rev. 180:177–89.
Article CAS PubMed Google Scholar - Caliezi C, et al. (2002) C1-inhibitor in patients with severe sepsis and septic shock: beneficial effect on renal dysfunction. Crit. Care Med. 30:1722–8.
Article CAS PubMed Google Scholar - Davis AE 3rd. (2006) Mechanism of angioedema in first complement component inhibitor deficiency. Immunol. Allergy Clin. North Am. 26:633–51.
Article PubMed Google Scholar - Fujii S, Hitomi Y. (1981) New synthetic inhibitors of C1r, C1 esterase, thrombin, plasmin, kallikrein and trypsin. Biochim. Biophys. Acta. 661:342–5.
Article CAS PubMed Google Scholar - Kobayashi T, Terao T, Maki M, Ikenoue T. (2001) Diagnosis and management of acute obstetrical DIC. Semin. Thromb. Hemost. 27:161–7.
Article CAS PubMed Google Scholar - Takahashi H, et al. (2003) Combined treatment with nafamostat mesilate and aspirin prevents heparin-induced thrombocytopenia in a hemodialysis patient. Clin. Nephrol. 59:458–62.
Article CAS PubMed Google Scholar - Ricklin D, Lambris JD. (2007) Complement-targeted therapeutics. Nat. Biotechnol. 25:1265–75.
Article CAS PubMed PubMed Central Google Scholar - Szalai AJ, et al. (2000) The Arthus reaction in rodents: species-specific requirement of complement. J. Immunol. 164:463–8.
Article CAS PubMed Google Scholar - Li JS, Jaggers J, Anderson PA. (2006) The use of TP10, soluble complement receptor 1, in cardiopulmonary bypass. Expert Rev. Cardiovasc. Ther. 4:649–54.
Article CAS PubMed Google Scholar - Rittershaus CW, et al. (1999) Recombinant glycoproteins that inhibit complement activation and also bind the selectin adhesion molecules. J. Biol. Chem. 274:11237–44.
Article CAS PubMed Google Scholar - Smith RA. (2002) Targeting anticomplement agents. Biochem. Soc. Trans. 30:1037–41.
Article CAS PubMed Google Scholar - De Silva RJ, et al. (2006) APT070 inhibits complement activation during in vitro cardiopulmonary bypass. Eur. J. Cardiothorac. Surg. 30:72–6.
Article PubMed Google Scholar - Sahu A, Lambris JD. (2000) Complement inhibitors: a resurgent concept in anti-inflammatory therapeutics. Immunopharmacology 49:133–48.
Article CAS PubMed Google Scholar - Chang ML, et al. (2009) Hepatic inflammation mediated by hepatitis C virus core protein is ameliorated by blocking complement activation. BMC Med. Genomics 2:51.
Article PubMed CAS PubMed Central Google Scholar - An F, et al. (2009) Role of DAF in protecting against T-cell autoreactivity that leads to experimental autoimmune uveitis. Invest. Ophthalmol. Vis. Sci. 50:3778–82.
Article PubMed Google Scholar - Donev RM, et al. (2008) Modulation of CD59 expression by restrictive silencer factor-derived peptides in cancer immunotherapy for neuroblastoma. Cancer Res. 68:5979–87.
Article CAS PubMed PubMed Central Google Scholar - Hill A, et al. (2006) Protection of erythrocytes from human complement-mediated lysis by membrane-targeted recombinant soluble CD59: a new approach to PNH therapy. Blood 107:2131–7.
Article CAS PubMed Google Scholar - Andersson J, Larsson R, Richter R, Ekdahl KN, Nilsson B. (2001) Binding of a model regulator of complement activation (RCA) to a biomaterial surface: surface-bound factor H inhibits complement activation. Biomaterials 22:2435–43.
Article CAS PubMed Google Scholar - Mikata S, et al. (1998) Regulation of complement-mediated swine endothelial cell lysis by a surface-bound form of human C4b binding protein. Transplantation 65:363–8.
Article CAS PubMed Google Scholar - Blom AM, Nandakumar KS, Holmdahl R. (2009) C4b-binding protein (C4BP) inhibits development of experimental arthritis in mice. Ann. Rheum. Dis. 68:136–42.
Article CAS PubMed Google Scholar - Hillmen P, et al. (2006) The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 355:1233–43.
Article CAS PubMed Google Scholar - Robak E, Robak T. (2009) Monoclonal antibodies in the treatment of systemic lupus erythematosus. Curr. Drug Targets 10:26–37.
Article CAS PubMed Google Scholar - Mache CJ, et al. (2009) Complement inhibitor eculizumab in atypical hemolytic uremic syndrome. Clin. J. Am. Soc. Nephrol. 4:1312–6.
Article CAS PubMed PubMed Central Google Scholar - Taube C, et al. (2006) Factor B of the alternative complement pathway regulates development of airway hyperresponsiveness and inflammation. Proc. Natl. Acad. Sci. U. S. A. 103:8084–9.
Article CAS PubMed PubMed Central Google Scholar - Gupta-Bansal R, Parent JB, Brunden KR. (2000) Inhibition of complement alternative pathway function with anti-properdin monoclonal antibodies. Mol. Immunol. 37:191–201.
Article CAS PubMed Google Scholar - Pawluczkowycz AW, et al. (2009) Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): considerably higher levels of CDC are induced by OFA than by RTX. J. Immunol. 183:749–58.
Article CAS PubMed Google Scholar - Castillo J, Milani C, Mendez-Allwood D. (2009) Ofatumumab, a second-generation anti-CD20 monoclonal antibody, for the treatment of lymphoproliferative and autoimmune disorders. Expert Opin. Investig. Drugs 18:491–500.
Article CAS PubMed Google Scholar - Woodruff TM, et al. (2006) Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. 20:1407–17.
Article CAS PubMed Google Scholar - Woodruff TM, et al. (2005) Increased potency of a novel complement factor 5a receptor antagonist in a rat model of inflammatory bowel disease. J. Pharmacol. Exp. Ther. 314:811–7.
Article CAS PubMed Google Scholar - Vergunst CE, et al. (2007) Blocking the receptor for C5a in patients with rheumatoid arthritis does not reduce synovial inflammation. Rheumatology (Oxford) 46:1773–8.
Article CAS Google Scholar - Ricklin D, Lambris JD. (2008) Compstatin: a complement inhibitor on its way to clinical application. Adv. Exp. Med. Biol. 632:273–92.
CAS PubMed PubMed Central Google Scholar - Holland MC, Morikis D, Lambris JD. (2004) Synthetic small-molecule complement inhibitors. Curr. Opin. Investig. Drugs 5:1164–73.
CAS PubMed Google Scholar - Valdimarsson H, et al. (2004) Human plasma-derived mannose-binding lectin: a phase I safety and pharmacokinetic study. Scand. J. Immunol. 59:97–102.
Article CAS PubMed Google Scholar - Gupta K, Gupta RK, Hajela K. (2008) Disease associations of mannose-binding lectin and potential of replacement therapy. Indian J. Med. Res. 127:431–40.
CAS PubMed Google Scholar - Mollnes TE, Kirschfink M. (2006) Strategies of therapeutic complement inhibition. Mol. Immunol. 43:107–21.
Article CAS PubMed Google Scholar - Ni Z, Hui P. (2009) Emerging pharmacologic therapies for wet age-related macular degeneration. Ophthalmologica 223:401–10.
Article CAS PubMed Google Scholar - Duvall MR, Hwang HY, Boackle RJ. (2010) Specific inhibition of the classical complement pathway with an engineered single-chain Fv to C1q globular heads decreases complement activation by apoptotic cells. Immunobiology. 215:395–405.
Article CAS PubMed Google Scholar - Arumugam TV, et al. (2004) Protective effect of a human C5a receptor antagonist against hepatic ischaemia-reperfusion injury in rats. J. Hepatol. 40:934–41.
Article CAS PubMed Google Scholar - Li Q, Nacion K, Bu H, Lin F. (2009) The complement inhibitor FUT-175 suppresses T cell autoreactivity in experimental autoimmune encephalomyelitis. Am. J. Pathol. 175:661–7.
Article CAS PubMed PubMed Central Google Scholar
Author information
Authors and Affiliations
- Department of Traumatology, Hand, Plastic, and Reconstructive Surgery, Center of Surgery, Center of Musculoskeletal Research, University of Ulm, Steinhövelstr. 9, 89075, Ulm, Germany
Christian Ehrnthaller, Florian Gebhard & Markus Huber-Lang - Institute of Orthopedic Research and Biomechanics, Center of Musculoskeletal Research, University of Ulm, Ulm, Germany
Anita Ignatius
Authors
- Christian Ehrnthaller
You can also search for this author inPubMed Google Scholar - Anita Ignatius
You can also search for this author inPubMed Google Scholar - Florian Gebhard
You can also search for this author inPubMed Google Scholar - Markus Huber-Lang
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toChristian Ehrnthaller.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ehrnthaller, C., Ignatius, A., Gebhard, F. et al. New Insights of an Old Defense System: Structure, Function, and Clinical Relevance of the Complement System.Mol Med 17, 317–329 (2011). https://doi.org/10.2119/molmed.2010.00149
- Received: 12 August 2010
- Accepted: 28 October 2010
- Published: 29 October 2010
- Issue Date: March 2011
- DOI: https://doi.org/10.2119/molmed.2010.00149