Signal Recognition Particle: An essential protein targeting machine (original) (raw)
. Author manuscript; available in PMC: 2013 Dec 2.
Abstract
The signal recognition particle (SRP) and its receptor comprise a universally conserved and essential cellular machinery that couples the synthesis of nascent proteins to their proper membrane localization. The past decade has witnessed an explosion in in-depth mechanistic investigations of this targeting machine at increasingly higher resolution. In this review, we summarize recent work that elucidates how the SRP and SRP receptor interact with the cargo protein and the target membrane, respectively, and how these interactions are coupled to a novel GTPase cycle in the SRP•SRP receptor complex to provide the driving force and enhance the fidelity of this fundamental cellular pathway. We also discuss emerging frontiers where important questions remain to be addressed.
Keywords: protein biogenesis, signal sequence, GTPases, SRP RNA, ribosome
INTRODUCTION
Proper localization of proteins to their correct cellular destinations is essential for sustaining the order and organization in all cells. Roughly 30% of the proteome are initially destined for the eukaryotic endoplasmic reticulum (ER), or the bacterial plasma membrane. Although the precise number of proteins remains to be determined, it is generally recognized that the majority of these proteins are delivered by the Signal Recongition Particle (SRP), a universally conserved protein targeting machine (1–4). Thirty years ago, the components and pathway for SRP-dependent protein targeting were first elucidated in mammalian cells through in vitro reconstitutions in cell extracts (5–9). The identification of the SRP homologue in prokaryotes a decade later further highlighted the salient, universally conserved features of this pathway (10–12). The biochemical accessibility of the bacterial SRP system has allowed for in-depth mechanistic investigations of this pathway, allowing us to understand its underlying molecular mechanism at unprecedented depth and resolution.
OVERVIEW OF SRP-DEPENDENT PROTEIN TARGETING
With the exception of the chloroplast SRP (see below), SRP-mediated protein targeting is a strictly cotranslational process that begins when a nascent polypeptide destined for the ER or plasma membrane emerges from the ribosome (Fig. 1A). The N-terminal signal sequence on the nascent polypeptide serves as the ‘signal’ that allows the ribosome•nascent chain complex (termed the RNC or cargo) to engage the SRP and, through interaction with the SRP receptor (SR), to be delivered to the vicinity of the Sec61p (or SecYEG in prokaryotes) translocon at the target membrane (Fig. 1A). There, the RNC is transferred to the Sec61p/SecYEG machinery, which either integrates the nascent polypeptide into the lipid bilayer or translocates it across the membrane to enter the secretory pathway. Meanwhile, SRP and SR dissociate from one another to mediate additional rounds of targeting (Fig. 1A).
Figure 1.

Overview of the pathways and components of SRP. (A) Multiple pathways deliver newly synthesized proteins to the ER or plasma membrane, with the SRP pathway mediating the co-translational targeting of translating ribosomes (right) and post-translational targeting machineries mediating the targeting of proteins released from the ribosome. (B) Domain structures of the ribonucleoprotein core of SRP, comprised of the SRP54 (or Ffh) protein and the SRP RNA (left), and the bacterial SRP receptor (right).
The size and composition of the SRP vary widely across species (see Fig. 5 and text below). Surprisingly, the bacterial SRP and SR, though highly simplified compared to those in eukaryotes, can replace their mammalian homologues to mediate efficient targeting of mammalian proteins to the ER (11, 13). This demonstrates the remarkable evolutionary conservation of the SRP pathway and shows that the functional core of SRP necessary and sufficient for protein targeting can be represented by the bacterial machinery. This provides a useful starting point for mechanistic dissections.
The bacterial SRP contains the universally conserved SRP54 protein (called Ffh in bacteria) bound to the 4.5S SRP RNA. Ffh has two structurally and functionally distinct domains (Fig. 1B, blue): a methionine-rich M-domain that recognizes the signal sequence and binds, with picomolar affinity, to the SRP RNA (14–16); and a special GTPase, NG-domain that interacts with a highly homologous NG-domain in the SR (17, 18) (Fig. 1B). The bacterial SR, called FtsY, also contains an N-terminal acidic A-domain that allows this receptor to peripherally associate with the membrane (19, 20).
The cotranslational SRP pathway minimizes the aggregation or misfolding of nascent proteins before they arrive at their cellular destination, and is therefore highly advantageous in the targeted delivery of membrane and secretory proteins. Nevertheless, an increasing number of post-translational protein targeting pathways have been identified (Fig. 1A, left). The best characterized thus far is the bacterial SecB/A system, which delivers bacterial secretory and outer-membrane proteins to the SecYEG complex and, through the ATPase cycles of SecA, drives the translocation of substrate proteins across the SecYEG translocon (1, 2). In yeast, the Sec62/63/71/72 system is a major pathway that mediates protein secretion (21, 22). Additional targeting pathways, including the Tat, Hsp70 and most recently the GET pathway, have been found (Fig. 1A, left path)(1, 2, 23–26).
Despite the divergence of targeting machineries, the SRP pathway illustrates several key features that are general to almost all protein targeting processes: (i) the cellular destination of a protein is dictated by its ‘signal sequence’, which allows it to engage a specific targeting machinery; (ii) targeting factors cycle between the cytosol and membrane, acting catalytically to bring cargo proteins to translocation sites at the target membrane; and (iii) targeting requires the accurate coordination of multiple dynamic events including cargo loading/unloading, targeting complex assembly/disassembly, and the productive handover of cargo from the targeting to translocation machinery. Not surprisingly, such molecular choreography requires energy input, which is often harnessed by GTPase or ATPase modules in the targeting machinery. Below, we discuss recent advances in our understanding of the molecular mechanisms that underlie these key events in the SRP pathway.
MOLECULAR INTERACTIONS AND REGULATION OF THE SRP CORE
Cargo Recognition by the SRP
Timely recognition of signal sequences by the SRP is essential for proper initiation of cotranslational protein targeting. Signal sequences that engage the SRP are characterized, in general, by a core of 8–12 hydrophobic amino acids that preferentially adopts an α-helical structure (27, 28). Crosslinking and phylogenetic analyses have implicated the M-domain of Ffh/SRP54 in binding the signal sequence (29–31). The unusually high methionine content of this domain further led to a ‘methionine bristle’ hypothesis, in which the flexible methionine side chains provide a hydrophobic environment with sufficient plasticity to accommodate a variety of signal sequences (10). In support of this model, crystallographic analyses of Ffh (16) and SRP54-signal peptide fusions (15, 32) showed that the signal sequence binds to a groove in the Ffh/SRP54 M-domain comprised almost exclusively of hydrophobic residues. Two different modes of signal peptide docking were observed (15, 32); this is probably due to the different signal sequences used in the two studies, and supports the notion that signal sequence interaction with the M-domain is quite flexible. A conserved, flexible fingerloop connects the α1 and α2 helices that line the ‘bottom’ of the signal sequence binding groove. This loop has been suggested to act as a flexible flap that closes upon the signal sequence (16, 33, 34), although direct evidence for this model remains to be obtained. Intriguingly, mutations in this loop disrupts the interaction between the SRP and SR GTPases (35), suggesting that it plays a role beyond that of facilitating signal sequence recognition. The precise role of the fingerloop remains to be clarified.
Despite these interactions, SRP binds isolated signal sequences weakly, with dissociation constants (_K_d) in the micromolar range (36, 37). In comparison, RNCs containing no signal sequences or even empty ribosomes bind the SRP with _K_d values of 80 –100 nM (38–40). Thus, the ribosome makes a significant contribution to the recruitment of SRP. The binding site of SRP on the ribosome was identified by crosslinking studies (41, 42) and cryo-electron microscopy (cryo-EM) reconstructions of the RNC•SRP complex in both the eukaryotic and bacterial systems (43–45). Together, these results show that basic residues on the ‘tip’ of the Ffh N-domain contact ribosomal proteins L23 and, to a lesser extent, L29 (Rpl25 and Rpl35 in eukaryotes, respectively) in the vicinity of the ribosome exit site (Fig. 2A). In the cryoEM structure, the M-domain also contacts ribosomal RNAs and perhaps ribosomal proteins L22 and L24, although these contacts remain to be verified biochemically. These ribosomal contacts, together with the interaction of the Ffh/SRP54 M-domain with the signal sequence, allow the SRP to bind its correct cargos with sub- to low-nanomolar affinity (38–40, 46).
Figure 2.
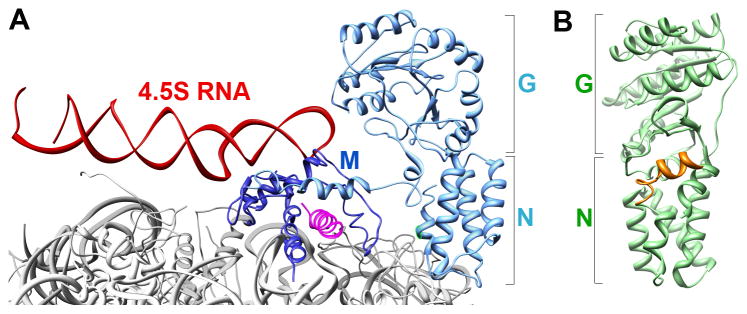
(A) Molecular model for interaction of the bacterial SRP with the translating ribosome (gray; PDB 2J28), derived from cryoEM reconstruction and docking of the crystal structures of individual protein fragments as described in (44). The M- and NG-domains of the SRP are in dark and light blue, respectively, the SRP RNA is in red, and the signal sequence is in magenta. (B) Crystal structure of the bacterial FtsY (NG+1) construct (PDB 2QY9) highlighting its lipid-binding helix at the N-terminus (orange).
Membrane localization of the SRP receptor
Bacterial SR is a single protein, FtsY, which lacks a bona fide transmembrane (TM) domain. The results of microscopy (47, 48), cell fractionation (49), and in vitro binding experiments using synthetic liposomes (19, 50, 51) indicate that the interaction of FtsY with the bacterial inner membrane is weaker and more dynamic than that of integral membrane proteins. Although the N-terminal A-domain has been speculated to mediate its membrane association, recent studies show that FtsY(NG+1), in which only Phe196 immediately preceding the NG-domain is retained, is sufficient to sustain lipid binding of FtsY and cotranslational protein targeting in vivo and in vitro (19, 51–53). Similar observations were made with the chloroplast FtsY homologue (54). Comparison of the crystal structure of FtsY(NG+1) with that of FtsY-NG (19, 55) showed that Phe196 induced folding of an amphiphilic α-helix rich in basic residues at the junction between the A- and N-domains, which provides FtsY’s primary lipid binding motif (Fig. 2B, orange).
This structure, together with in vitro binding studies, also showed that FtsY preferentially binds anionic phospholipids, phosphatidylglycerol (PG) and cardiolipin (CL) (19, 50, 51). This preference is corroborated by experiments in vivo, in which an FtsY mutant defective in lipid binding was rescued by overexpression of genes involved in PG and CL biosynthesis (56). Anionic phospholipids have also been found to preferentially interact with and activate the SecYEG machinery (57) and the SecA ATPase (58, 59), and stimulate the integration and export of membrane and secretory proteins (60–62). Together, these observations suggest that sites of bacterial inner membrane enriched in anionic phospholipids could constitute active zones for protein targeting and translocation, an attractive hypothesis that remain to be tested.
In addition to lipid interactions, a direct interaction of FtsY with SecYEG would provide an attractive mechanism to more precisely localize the targeting complex to the translocon. Evidence for this interaction was obtained recently in crosslinking and co-purification studies (63, 64). Subsequent crosslinking and mutational studies further showed that the A-domain of FtsY and the cytosolic loops of SecYEG connecting TMs 6–7 and TMs 8–9 (termed C4 and C5 loops in prokaryotes and L6/7 and L8/9 loops in eukaryotes) participate in this interaction (20, 64, 65). Nevertheless, several puzzling observations remain unexplained. Given the low sequence conservation of the FtsY A-domain and its dispensability for cotranslational targeting, it is unclear to what extent this domain helps facilitate the targeting reaction. The NG-domain of FtsY was also suggested to interact with SecYEG (65), but direct evidence for this interaction remains to be obtained. Most importantly, the SecYEG C4 and C5 loops that interact with FtsY are also crucial for its interaction with the ribosome ((65); see section below), suggesting that the interaction of FtsY with SecYEG is transient and must be broken to allow for stable docking of RNC onto the SecYEG machinery. The timing, mechanism, and precise roles of the FtsY-SecYEG interaction remain challenging questions for future studies.
Eukaryotic SR is a heterodimeric complex comprised of the α and β subunits (66). SRα is a soluble protein highly homologous to FtsY. Instead of the A-domain, SRα contains an N-terminal X-domain that dimerizes with SRβ, an integral membrane protein, thus localizing the SRP receptor to the ER membrane (67). SRβ also contains a GTPase domain that, unlike the two GTPases in SRP and FtsY/SRα described later, is most homologous to the Arf family of GTPases (67, 68). Intriguingly, stable SRα–β association requires SRβ to be bound with GTP (67), and the Sec61β subunit of Sec61p complex could accelerate GDP dissociation from SRβ (69), suggesting that Sec61β potentially serves as a nucleotide exchange factor that maintains SRβ in the GTP-bound state active for binding SRα. Direct interaction of SRβ with the yeast Sec61p homologue, Ssh1p, was demonstrated in vivo using a split ubiquitin assay (70), and disruption of this interaction impairs cotranslational protein targeting and cell growth (71). These results suggest functional interactions of the eukaryotic SR with the Sec61p translocon that parallel findings with the bacterial FtsY, and show that the membrane localization of the eukaryotic SR may be subject to more complex regulation.
Regulation of protein targeting by the SRP and SRP receptor GTPases
At the membrane, SRP and SR meet and interact with one another through their GTPase modules. Both proteins contain a central GTPase, G-domain that shares homology with the classic P-loop GTPase fold (55, 72). Unique to the SRP and SR GTPases is an additional β–α–β–α insertion box domain (IBD), in which a flexible IBD loop (red in Fig. 3A) contains multiple catalytic residues and provides an equivalent of the switch II loop in _Ras-_type GTPases (55, 72). In addition, a four helix bundle preceding the GTPase fold forms the N-domain, which together with the G-domain comprises a structural and functional unit termed the NG-domain (Fig. 2). Unlike classic signaling GTPases that exert regulation by switching between a GTP-bound, active state and a GDP-bound, inactive state (73, 74), SRP and SR represent a novel class of GTPases whose activities are regulated by nucleotide-dependent dimerization cycles (75). Members of this family also include FlhF, MinD, MnmE, the Septins, Toc proteins, human guanylate binding protein-1, and the dynamin family of GTPases (75–78). In the past decade, mechanistic studies of the bacterial SRP and SR GTPases have elucidated the biological logic and regulatory mechanism for these twin GTPases, which could provide general principles for understanding other members of this GTPase family.
Figure 3.

Conformational changes in the SRP and SR GTPases ensure the efficiency and fidelity of protein targeting. The steps are numbered to be consistent between parts (A) and (B). The Ffh and FtsY NG domains are in blue and green, respectively. T and D denote GTP and GDP, respectively. (A) A series of discrete rearrangements drive the SRP•SR GTPase cycle and are regulated by the cargo and target membrane. ⊥ denotes the pausing effect of cargo in disfavoring the conformational rearrangements. Right panel: molecular model of the early intermediate (PDB 2XKV). Bottom panel: Co-crystal structure of the Ffh-FtsY NG-domain complex in the closed/activated conformation (PDB 1RJ9). The two GTP analogues are in spacefill. Left panel: Zoom-in of the composite active site formed at the dimer interface required for GTPase activation, with the GMPPCP molecules from Ffh and FtsY in blue and green, respectively, active site Mg2+ in magenta, nucleophilic waters (W) in blue, and catalytic residues in the IBD loops in red. (B) GTPase rearrangements provide the driving force and ensure the fidelity of protein targeting. Step 1, RNC with a signal sequence (magenta) binds the SRP. Step 2, cargo-loaded SRP forms a stabilized early targeting complex with FtsY. Step 3, membrane association of FtsY drives rearrangement to the closed state, which weakens SRP’s affinity for the cargo. Step 4, interaction of SR with the SecYEG translocon is proposed to drive GTPase rearrangements to the activated state required for cargo handover. Step 5, the cargo is unloaded from the SRP onto SecYEG, and GTP hydrolysis drives the disassembly and recycling of SRP and SR. At each step, the cargo can be either retained in (black arrows) or rejected from (red arrows) the SRP pathway.
Free Ffh and FtsY exhibit minor structural differences amongst the apo, GDP-, and GTP-bound states (55, 72, 79–82). Even with GTP bound, both GTPases by themselves are in an inactive open conformation, exhibiting fast nucleotide dissociation and exchange rates as their nucleotide binding pocket is wide-open (Fig. 2 and 3A), and low basal GTPase activity as their catalytic loops are not correctly positioned (83). Their GTPase cycle is driven by a series of conformational changes during their dimerization that culminate in reciprocal GTPase activation (Fig. 3; (84)). GTPase assembly is initiated with a transient early intermediate, which forms rapidly but is highly unstable (Kd ~ 4–10 μM; Fig. 3, step 2)(85). This intermediate lacks stable contacts between the G-domains of Ffh and FtsY, and is primarily stabilized by electrostatic attractions between their N-domains (Fig. 3A, right panel)(86, 87). Subsequent GTP-dependent rearrangements, primarily involving readjustments at the intra-molecular G-N domain interface (17, 18, 88, 89) and removal of an inhibitory N-terminal helix (90–92), lead to the formation of a stable closed complex in which extensive stereospecific interactions are formed between G-domains of both proteins (Kd ~16 – 30 nM; Fig. 3A, step 3 and lower panel). Two pairs of hydrogen bonds are formed across the dimer interface through the 3′-OH of one GTP and the γ-phosphoryl oxygen of the other, which further stabilize the GTPase dimer (17, 18). The final GTPase activation step involves local rearrangements of the IBD loops, which must be brought into close proximity to the GTP molecules to form an activated complex (Fig. 3A, step 4). Each IBD loop provides at least three catalytic residues (Asp135, Arg138, and Gln148 in Ffh and their homologous residues in FtsY) that coordinate the nucleophilic water, the γ-phosphoryl oxygen and the active site Mg2+, forming a composite active site conducive to hydrolyzing GTP (Fig. 3A, left panel)(17, 18, 89). Following hydrolysis, the SRP•FtsY complex is much less stable and quickly disassembles (Fig. 3A, step 5; (83, 93)).
Importantly, each of the GTPase rearrangements during the dimerization and activation of SRP and FtsY provides a discrete regulatory point at which they can sense and respond to the presence of the RNC and target membrane, thus allowing the loading of cargo on the SRP to be tightly coupled to its delivery to the membrane. For example, with free SRP and FtsY, assembly of a stable closed complex is extremely slow (_k_on ~102–103 M−1s−1; (36, 83, 94)) and insufficient to sustain the protein targeting reaction. The RNC, by stabilizing the early intermediate over 100-fold and preventing its premature disassembly, accelerates stable SRP•FtsY assembly 103-fold (95). Analogously, phospholipid membranes, by helping to pre-organize FtsY into the closed conformation, accelerate GTPase assembly 160-fold (51, 96, 97). These allosteric regulations ensure the rapid delivery of cargo to the membrane, and minimize futile cycles of interactions between the free SRP and SR.
Intriguingly, the RNC also disfavors the rearrangement of the GTPase complex to the closed and activated states, and delays GTPase activation in the targeting complex (40, 95). This generates a highly stabilized early targeting intermediate in which the RNC is predicted to bind the SRP with picomolar affinity, while GTP hydrolysis is ‘paused’ (95). These effects are highly beneficial in preventing abortive reactions at early stages of targeting; however, they pose serious challenges for the cargo unloading and GTPase activation events at later stages. Multiple observations strongly suggest that the resolution to this problem is provided in part by the subsequent GTPase rearrangements to the closed and activated states, which helps switch the targeting complex from a cargo-binding to cargo-releasing mode. The interaction of cargo with SRP is estimated to weaken ~400-fold when the early targeting complex rearranges to the subsequent conformational states (95). Further, mutant GTPases that block the closed → activated rearrangement specifically block the engagement of cargo with the translocon (98). Finally, crosslinking and cryo-EM analyses showed that in the presence of SR and GTP analogues, the NG-domain of SRP becomes mobile and detaches from L23 (42, 99). Importantly, these late GTPase rearrangements can be induced by anionic phospholipid membranes (Fig. 3, step 3; (51)), suggesting an attractive mechanism to spatially couple the membrane delivery of RNCs to their subsequent unloading.
Collectively, these results provide a coherent picture for how the unusual GTPase cycle of SRP and SR is used to provide exquisite spatial and temporal coordination of protein targeting (Fig. 3B). GTPase assembly is minimized in the absence of biological cues, but is initiated when the SRP is loaded with RNCs bearing strong signal sequences (steps 1–2). In the absence of the target membrane, however, the RNC•SRP•SR complex is primarily stalled in the early conformational stage, where the cargo is tightly bound to the SRP and GTP hydrolysis is delayed. Interaction of FtsY with phospholipid membranes helps relieve this ‘pause’ and induce the GTPase rearrangements into the closed/activated states, in which the interaction of the ribosome with the SRP is weakened and the RNC could be more readily released from the targeting complex (step 3). It is still unclear what ultimately drives the completion of the cargo handover event and reactivates GTP hydrolysis (steps 4–5), although the SecYEG translocon provides an attractive candidate. Finally, GTP hydrolysis drives the disassembly and recycling of SRP and SR, allowing them to initiate new rounds of protein targeting.
Fidelity of SRP: binding, induced fit, and kinetic proofreading
Like other topogenic sequences that mediate protein localization, SRP signal sequences are highly divergent (27, 28, 100, 101), and the SRP must be sufficiently flexible to accommodate this diversity. Nevertheless, SRP must also remain highly specific to its substrates to minimize the mislocalization of proteins, which would be detrimental to cells. How the SRP or any protein targeting machinery faithfully selects their correct substrates has been a challenging question. Although previous work has focused on the observation that SRP binds weakly to the ‘incorrect’ cargos bearing no or weak signal sequences (Figure 3B, red arrow a), quantitative biophysical measurements show that SRP binds with substantial affinity to the incorrect cargos or even the empty ribosome (Kd ~ 80–100 nM; (38–40)). Given the cellular SRP concentration (~400 nM in bacteria), it appears unlikely that the discrimination in the cargo-binding step is sufficient to reject all the incorrect cargos.
A quantitative dissection of the individual molecular events in the bacterial SRP pathway (Fig. 3B) demonstrates that the multiple conformational rearrangements in the SRP•FtsY GTPase complex provide a series of additional checkpoints to further reject the incorrect cargos (40). These include: (i) formation of the early intermediate, which is stabilized over 100-fold by the correct, but not incorrect cargos (Figure 3B, red arrow b); (ii) rearrangement of the early intermediate to the closed complex, which is ~10-fold faster with the correct than the incorrect cargos (Figure 3B, red arrow c); and (iii) GTP hydrolysis by the SRP•FtsY complex, which is delayed ~8-fold by the correct cargo to give the targeting complex a sufficient time window to identify the membrane translocon. In contrast, GTP hydrolysis remains rapid with the incorrect cargo (_t_1/2 < 1s), which could abort the targeting of incorrect cargos (Figure 3B, arrow d). A mathematical simulation based on the kinetic and thermodynamic parameters of each step strongly suggest that all these fidelity checkpoints are required to reproduce the experimentally observed pattern of substrate selection by the SRP (40).
These results support a novel model in which the fidelity of protein targeting by the SRP is achieved through the cumulative effect of multiple checkpoints, by using a combination of mechanisms including cargo binding, induced SRP–SR assembly, and kinetic proofreading through GTP hydrolysis. Additional discrimination could be provided by the SecYEG machinery, which further rejects the incorrect cargos (102). Analogous principles have been demonstrated in the DNA and RNA polymerases (103, 104), the spliceosome (105), tRNA synthetases (106) and tRNA selection by the ribosome (107), and may represent a general principle for complex biological pathways that need to distinguish between the correct and incorrect substrates based on minor differences.
SRP RNA: a central regulator of the SRP
Besides the SRP54 (or Ffh) protein, the SRP RNA is the only other universally conserved and essential component of the SRP (108). However, its precise roles in protein targeting have remained enigmatic. In early biochemical reconstitutions of the mammalian SRP, the SRP RNA appeared nothing more than a scaffold that holds different SRP protein subunits together (Fig. 5 below). The discovery of the bacterial SRP RNA (109), which binds a single protein Ffh, implied a much more active role for this RNA. Recent biochemical and structural studies strongly support this view and show that the SRP RNA can mediate global reorganization of the SRP in response to cargo binding and provide additional interactions with the SR, thus mediating the molecular communication between the cargo and the SRP/SR GTPases during protein targeting.
The bacterial 4.5S SRP RNA contains the most conserved domain IV of the SRP RNA and forms an elongated hairpin structure capped by a highly conserved GGAA tetraloop at one end (Fig. 4A). Two internal loops, A and B, mediate binding of this RNA to a helix-turn-helix motif in the M-domain of Ffh with picomolar affinity (14, 110). The orientation of the M-domain/RNA complex relative to the Ffh NG-domain, however, exhibits a high degree of variability. Crystallographic analyses and structural mapping studies have generated at least four different structures or structural models of the SRP, each exhibiting a distinct inter-domain arrangement (Fig. 4B for two examples; (16, 33, 34, 111–113)). Collectively, these observations suggest that apo-SRP could exist in a variety of global conformations, likely due to the 30 amino acid-long flexible linker connecting the M- and NG-domains of Ffh.
Figure 4.
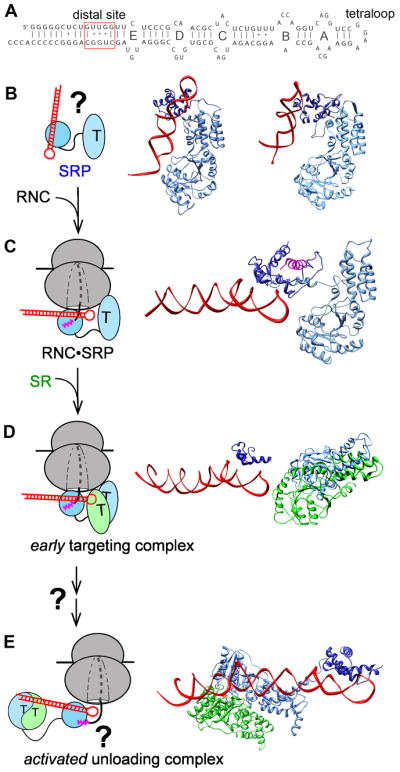
RNA-mediated global reorganization of the SRP couples the GTPase cycle to the cargo loading and unloading events during protein targeting. (A) Secondary structure of the E. coli 4.5S SRP RNA. The internal loops A–E, the GGAA tetraloop and the distal site near the 5′,3′-end of this RNA are denoted. (B) Free SRP exist in a variety of ‘latent’ conformations in which the SRP RNA tetraloop is not positioned to contact SR. Two representative structures of SRP from Methanococcus jannaschii (left; PDB 2V3C) and Sulfolobus solfataricus (right; PDB 1QZW) are shown. (C) Binding of the RNC induces SRP into a more active conformation, in which the SRP RNA tetraloop is properly positioned to interact with the G-domain of the incoming SR to form a stabilized early targeting complex in (D). Both panels show the molecular model derived from cryo-EM reconstructions of the RNC•SRP or RNC•SRP•FtsY early complex; the ribosome is not shown for clarity. (E) GTPase activation is potentially coupled to relocalization of the SRP•SR NG-domain complex to the distal end of the SRP RNA, a conformation that is more conducive to cargo unloading (PDB 2XXA). The structures in (B) and (C) are aligned with respect to the SRP54-NG domain, and the structures in (C) – (E) are aligned with respect to the SRP RNA. Color codings are the same as in Figure 2.
Upon binding the RNC, however, the SRP undergoes a global conformational change (Fig. 4C; (44, 45, 114)). The bi-dentate interaction of the RNC with Ffh re-orients its M- and NG-domains, such that the SRP RNA now lies parallel to the ribosome surface, with its GGAA tetraloop positioned adjacent to the FtsY-interacting surface on the Ffh NG-domain (Fig. 4C). This is important, as the RNA tetraloop is required for rapid assembly of a stable SRP•FtsY complex (83, 85, 94, 115, 116). More recent kinetic and phylogenetic analyses (117), hydroxylradical footprinting experiments (118), and cryoEM analysis (86) identified a key electrostatic interaction between the SRP RNA tetraloop and conserved basic residues surrounding Lys399 on the lateral surface of FtsY (Fig. 4D). This interaction stabilizes the otherwise highly labile early intermediate, thus accelerating stable SRP•FtsY assembly 102–103 fold (85, 117). Importantly, the RNA tetraloop or FtsY Lys399 exerts these stimulatory effects only when the SRP is bound to RNCs bearing strong signal sequences (117, 119) and, to a lesser extent, to signal peptide or signal peptide mimics (36). Combined with structural analyses (32, 45, 86, 99), a coherent model emerges in which the RNC optimizes the conformation of SRP so that the SRP RNA tetraloop is pre-positioned to interact with the incoming FtsY, thus allowing rapid recruitment of the SR to be achieved specifically for the correct cargos (Fig 4B – D).
Intriguingly, neither the SRP RNA tetraloop nor FtsY Lys399 affect the equilibrium stability of the SRP•FtsY complex in the closed/activated states (94, 117), suggesting that their interaction is highly transient and occurs only during the early intermediate stage of GTPase dimer assembly. A recent crystallographic study using full-length 4.5S RNA (120) revealed a completely different configuration of the SRP•FtsY complex, in which a closed/activated GTPase complex docks at a distinct site near the 5′, 3′-end of the SRP RNA ~100 Å away from the tetraloop end (Fig. 4A, distal site and Fig. 4E). Mutations of the distal site compromised GTPase activation in the SRP•FtsY complex, supporting the importance of this alternative RNA-GTPase interaction (120). Although it is still at early stages, these results suggest an intriguing model in which the Ffh•FtsY NG-domain complex, after initial assembly near the RNA tetraloop, relocalizes to the opposite end of the SRP RNA where its GTPase activity is fully activated. In the context of the protein targeting reaction, this movement is highly attractive as it removes the GTPase complex from the ribosome exit site, generating a conformation that allows the RNC to be more easily released from the targeting complex and the SecYEG complex to more readily access the ribosome exit site (Fig. 4E). In addition, the unloading of cargo could be tightly coupled to GTPase activation in such a mechanism. Nevertheless, direct evidence for such a largescale GTPase movement on the SRP RNA, its underlying driving forces and molecular mechanisms, and its precise roles in the protein targeting reaction remain to be demonstrated.
Transition from the targeting to translocation machinery
At the end of the protein targeting reaction, the RNC must be unloaded from the SRP•FtsY complex onto the heterotrimeric SecYEG (or Sec61p) translocation machinery. The readers are referred to (2, 121–123) for more comprehensive reviews of this machinery. In the context of the cotranslational targeting reaction, rich structural information has been obtained in recent years to explain how the translocon interacts with the RNC and potentially interfaces with the SRP targeting machinery. A crystal structure of M. jannaschii SecYEβ (124) showed that TMs 1–10 of SecY form an hourglass shaped pore in this channel. Lining one side of this pore are TMs 2b and 7, which form the lateral gate where hydrophobic signal sequences and TMs in the nascent polypeptide bind and subsequently enter the lipid bilayer (125–127). Cryo-EM reconstructions of the RNC•SecYEG complex (or its eukaryotic homologues) at increasing resolution (128–131), combined with biochemical and genetic studies (132, 133), further identified the highly conserved basic residues in the C4 and C5 (or L6/7 and L8/9) loops of SecY as the key motifs that mediate interaction with ribosomal proteins L23 and L35 at the exit site.
Remarkably, the binding sites of the SecYEG/Sec61p complex on the translating ribosome overlap extensively with those of the SRP (Fig. 2A). This raises challenging questions as to how the RNC is handed over from the targeting to translocation machinery without nonproductive loss of the translating ribosome. The most productive mechanism for the cargo transfer event is probably through a concerted pathway, in which the two contacts of SRP with the RNC, those with the L23/L35 ribosomal proteins and with the signal sequence, are sequentially dissolved and replaced by those of the SecYEG machinery. Many observations described earlier, including the loss of density for the Ffh-FtsY NG-domain complex in cryoEM reconstructions of the targeting complex (99), the ability of the NG-domain complex to relocalize to the SRP RNA distal end (120), and the requirement of GTPase rearrangements for detachment of SRP from the ribosome (95, 98) provide clues that support such a mechanism. The ability of the SR to directly interact with the SecYEG/Sec61p complex (64, 65, 69–71) further raises the possibility that the translocon plays an active role in the cargo handover process. Nevertheless, the cargo handover event remains the least understood aspect of the cotranslational targeting reaction. The fate of the ribosomal proteins and the signal sequence in this cargo handover event, their timing relative to one another and to the hydrolysis of GTP, and the molecular forces that drive this step remain challenging questions for future investigations.
EUKARYOTIC SRP
Mammalian SRP: additional layers of complexity
Compared to its bacterial homologue, the mammalian SRP is significantly larger and more complex, comprised of six proteins and a 7S SRP RNA (Fig. 5). It can be divided into two distinct domains: the S-domain, comprised of domains II–IV of the 7S RNA and the SRP 19, 54, and 68/72 protein subunits, and the Alu domain, comprised of domain I of the 7S RNA and the SRP 9/14 subunits (Fig. 5). The increased complexity adds additional layers of nuance and regulation for the mammalian SRP, many of which await to be elucidated.
Figure 5.

Organization of the mammalian SRP. (A) Comparison of the RNA secondary structure and composition of the mammalian and bacterial SRP. The SRP54 M- and NG-domains are in dark and light blue, respectively, SRP19 is in cyan, SRP9 is in brown, SRP14 is in orange, and the SRP68/72 complex, which lacks a crystal structure, is represented as a gray sphere. (B) Cryo-EM reconstruction of the mammalian SRP bound to the RNC (left; EMD-1063), and molecular model of the mammalian SRP derived from the cryo-EM and docking of the crystal structures of the individual proteins (right; PDB 1RY1). The S- and Alu-domains of the SRP RNA are in red and yellow, respectively, the protein subunits are colored as in (A).
For example, the mammalian SRP54 subunit binds the 7S RNA weakly by itself. Indeed, premature binding of SRP54 could cause the two RNA-binding loops for SRP19 to misfold, disrupting the native assembly of SRP (134, 135). In vivo, assembly of the mammalian SRP goes through an ordered pathway in which all the SRP proteins except SRP54 are imported to the nucleus to bind the SRP RNA; the partially assembled SRP is then exported to the cytoplasm for SRP54 binding, thus completing its assembly [(109, 136–138); see (139) for a more complete review of SRP assembly]. In vitro reconstitutions showed that pre-binding of SRP19 to the 7S RNA is required for loading the SRP54 subunit onto the SRP RNA (8, 140). Crystallographic analyses showed that SRP19 bridges the two tetraloops in both domains III and IV (or helices 6 and 8) of the 7S RNA and preorganizes the internal loops in domain IV into a conformation required for stable SRP54 binding [(141–146); see (147) for a more complete review]. Why the mammalian SRP requires this additional layer of allostery during its assembly remains unclear.
In addition, although much is known about the binding sites of SRP68/72 on the 7S RNA (148–153), the structure and precise function of the SRP68/72 subunits remain to be defined. Chemical probing experiments have suggested that SRP68/72 cooperates with SRP19 to preorganize the 7S RNA into a conformation competent for SRP54 binding, by exposing the SRP54 binding sites on the 7S RNA (144, 154). These subunits have also been implicated in controlling the interaction of SRP54 with the SR (155). Direct evidence for both of these models remains to be obtained.
The most interesting aspect of the mammalian SRP, aside from the core functions, is the ‘Alu’ domain (Fig. 5) that arrests translation elongation just after the signal sequence emerges from the ribosome. Early biochemical work found that SRP interacts with the ribosome during elongation factor-2 catalyzed translocation of tRNA (156), suggesting that it competes with elongation factors for binding. Recent biochemical and crosslinking studies further show that SRP9/14 electrostatically interacts with ribosomal RNA via at least two stretches of basic residues, and also contacts ribosomal proteins at the interface between the large and small ribosomal subunits (157, 158). Consistent with this notion, cryo-EM analysis showed that mammalian SRP forms an elongated, kinked structure in which the Alu domain reaches into the elongation factor binding site at the ribosome subunit interface (43) (Fig. 5B). Although the elongation arrest activity is not a prerequisite for protein targeting in vitro, deletion of SRP9/14 in vivo results in severe defects in protein targeting and mammalian cell growth (159). Together with the observation that the SRP could not target proteins when the nascent polypeptide exceeds a critical length (39, 160), these results suggest that elongation arrest provides a crucial time window that allows the targeting complex to engage the translocon before the nascent chain loses translocation competence. The precise mechanism and degree of elongation arrest by the Alu-domain, and how it communicates and/or cooperates with the S-domain during the targeting reaction remain to be determined.
Chloroplast SRP: a unique post-translational SRP
The cotranslational nature of the SRP pathway is universally conserved except for the chloroplast in green plants, where a unique post-translational SRP pathway has evolved. Instead of delivering RNCs as its cargo, the chloroplast SRP (cpSRP) is dedicated to the delivery of the light harvesting chlorophyll-binding proteins (LHCP) from the chloroplast stroma to the thylakoid membrane (Fig. 6)(161, 162). Analogous to the cytosolic SRP, the cpSRP pathway is mediated by close homologues of the SRP54 and SR GTPases, cpSRP54 and cpFtsY, respectively (Fig. 6) (162–165). However, the cpSRP54 M-domain lost the ability to bind the otherwise universally conserved SRP RNA (166). Instead, a unique SRP subunit in chloroplast, cpSRP43, binds a C-terminal extension in the cpSRP54 M-domain to form the cpSRP (Fig. 6)(167, 168). As detailed below, these changes likely reflect adaptation of the cpSRP system to the post-translational targeting of LHCPs. In addition, another pool of cpSRP43-free cpSRP54 was found in stroma, which together with cpFtsY mediate the cotranslational targeting of some of the chloroplast-encoded membrane proteins, such as D1 (169). The readers are referred to (170–172) for comprehensive reviews of the cpSRP. Here, we will focus on valuable lessons that came from comparison of the cpSRP with the classic cytosolic SRP in recent years.
Figure 6.
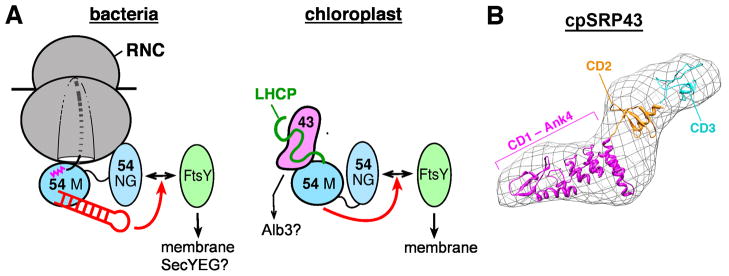
(A) Similarity and differences between the bacterial (left) and chloroplast (right) SRP systems. The SRP54 M- and NG-domains, FtsY, and the SRP RNA are colored as in Figure 2. The LHC protein (LHCP) is in green, and cpSRP43 is in magenta. The red arrows denote stimulatory effects of the SRP RNA (left) and the cpSRP54 M-domain (right) on assembly of the GTPase complex. (B) A molecular model of cpSRP43, obtained from small angle x-ray reconstructions of its 3-D shape (envelope; (183)) and rigid body docking of the structures of the CD1-Ank(1-4)-CD2 (PDB 3UI2) and CD3 (PDB 1X3P) fragments.
How does the cpSRP bypass the otherwise strictly conserved SRP RNA? In cytosolic systems, a major function of the SRP RNA is to accelerate the interaction between the SRP and FtsY GTPases and thus ensure the rapid delivery of cargo. Kinetic analysis in the cpSRP system showed that, even in the absence of the SRP RNA, the cpSRP and cpFtsY GTPases interact 400-fold faster than their bacterial homologues (173). Subsequent crystallographic (174, 175) and biochemical cross-complementation (176) analyses revealed two key molecular mechanisms underlying this phenomenon: (i) compared to bacterial FtsY, the conformation of the cpFtsY NG-domain more closely resembles that in the closed SRP•FtsY complex; this may allow cpFtsY to bypass some of the rearrangements required for stable GTPase assembly (174, 175); (ii) more importantly, the cpSRP54 M-domain functionally mimics the SRP RNA, accelerating its interaction with cpFtsY 100-fold and allowing them to achieve an interaction rate that matches the RNA-catalyzed interaction between their bacterial homologues (Fig. 6, red arrows)(176). It is probable that analogous to the cytosolic SRP system, the interaction between the cpSRP and cpFtsY GTPases are regulated by upstream and downstream components of the pathway, such as the substrate protein or the target membrane (177); these allosteric regulations and their roles in the cpSRP pathway remain to be uncovered.
The cpSRP43 subunit is responsible for substrate recognition and enables the cpSRP to adapt to the challenge of post-translational protein targeting. Unlike in the cotranslational pathway, cpSRP must handle fully synthesized, highly hydrophobic LHCPs that are prone to aggregation and misfolding in aqueous environments. Early work found that LHCPs are effectively chaperoned in the stroma where they form a soluble ‘transit complex’ with the cpSRP (162, 164, 178–180), although substrate capture by the cpSRP may require additional factors, such as LTD at the chloroplast envelope (181). Recent biochemical dissections showed that cpSRP43 is necessary and sufficient for binding with high affinity to LHCPs and maintaining them in a soluble, translocation competent state (Fig. 6, right)(182, 183). cpSRP43 is comprised of a unique combination of protein-interaction motifs, with three chromodomains [CDs; (184, 185)] and four ankyrin (Ank) repeats (Ank1–4) sandwiched between CD1 and CD2 (Fig. 6B) (172, 186). The ankyrin repeat domain specifically recognizes L18, a relatively polar 18-amino acid motif between TM2 and TM3 of LHCP (180, 187, 188). Crystallographic analyses further showed that the CD1-Ank4 fragment of cpSRP43 folds into an elongated horseshoe structure (Fig. 6B), in which a groove across the concave surface of Ank2 to 4 binds a highly conserved DPLG turn in the L18 peptide (189), enabling specific recognition of LHCPs by cpSRP43. As a molecular chaperone, cpSRP43 likely also interacts with and shields the hydrophobic TMs in LHCPs, although the molecular basis of these interactions remains to be deciphered. Finally, recent work found that even after LHCPs already aggregated, cpSRP43 can resolubilize the aggregate and return them to soluble fractions in vitro (182, 183). This ‘disaggregase’ activity was unexpected, as cpSRP43 lacks ATPase domains and hence must use a mechanism distinct from that of the well-studied AAA+ disaggregase systems (190). This finding demonstrated the capability and diversity of chaperone function during post-translational membrane protein targeting. The molecular basis underlying cpSRP43’s ‘disaggregase’ activity and its precise roles in LHCP biogenesis in vivo remain to be determined.
At the thylakoid membrane, LHCPs are delivered by the cpSRP and cpFtsY to the Alb3 translocase (see more discussion of this translocase later). Recently, a direct interaction between cpSRP43 and the C-terminal stromal domain of Alb3 has been shown in biochemical studies (191, 192) and in vivo complementation analyses (193, 194). The molecular mechanism underlying this interaction and its precise roles in the targeting and integration of LHCP remain to be clarified. Nevertheless, this interaction is highly attractive, as it provides a mechanism to accurately localize the targeting complex to the Alb3 translocase and to couple the membrane delivery of LHCP to its subsequent integration. Lessons learned from this system could be leveraged to help understand the mechanism of cargo unloading in the cytosolic SRP pathway.
NEW FRONTIERS
Molecular code of the signal sequence
Early pioneering work has identified a hydrophobic core as the major determinant of signal sequences that mediate protein secretion, facilitated by basic amino acids at the N-terminus in some cases (27, 28). The propensity to adopt α-helical structures in apolar media has also been identified as an important determinant of the signal sequence (195, 196). However, subsequent work revealed additional layers of complexity. First, multiple pathways mediate protein secretion in bacteria and yeast, and signal sequences also specify the targeting pathway (Fig. 1A)(101). Second, although a threshold level of hydrophobicity in signal sequences was generally thought to specify the SRP pathway, more recent studies in bacteria (197) and yeast (198) indicated that the correlation between hydrophobicity and SRP-dependent targeting is poor, and signal sequences with hydrophobicity above the apparent ‘threshold’ failed to engage the SRP (197). Third, special N-extensions of a strong SRP signal sequence, such as those found in the bacterial autotransporter EspP, can allow nascent proteins to escape the SRP pathway (40, 199). Apparently, additional molecular features of the signal sequence play important roles, including helical propensity (195, 196), the presence of N-terminal basic residues (28, 200), and additional properties that have yet to be identified. How the information from all the different features is integrated to comprise the ‘molecular code’ that specify the SRP remains unclear. Crucial to the effort to ‘decode’ the signal sequence will be the availability of a more comprehensive catalogue of validated SRP-dependent vs. SRP-independent substrates, which would allow more systematic analyses of the molecular features of signal sequences and evaluation of their respective contributions to recognition by the SRP.
The crowded ribosome exit site
Accumulating data now indicate that the ribosome exit site is a crowded environment where multiple protein biogenesis factors interact. As a newly synthesized protein emerges from the ribosomal exit tunnel, it interacts with a host of cellular factors that facilitate its folding, localization, maturation, and quality control. These include molecular chaperones such as trigger factor (TF) in bacteria, Hsp70 (DnaK/J in bacteria), and the nascent chain-associated complex (NAC) in yeast; modification and processing enzymes such as methionine aminopeptidase (or peptide deformylase in bacteria), N-acetyl transferase, and arginyl transferase; and protein targeting and translocation machineries such as the SRP and SecYEG (1, 201, 202). Even post-translational targeting factors, such as SecA (203) and the Bag6 complex (204), were recently reported to interact with the RNC. Many of these factors, including the SRP, SecYEG, TF and SecA, contact the ribosome via the same protein, L23 (or Rpl25 in eukaryotes) (205), and recognize hydrophobic sequences on the nascent polypeptide. It is currently unclear whether and how these factors compete or collaborate with one another for binding the translating ribosome (198, 206–211). Further, the molecular mechanisms by which a nascent protein is sorted among different cotranslational factors and committed to the correct biogenesis pathway remain key questions to be addressed in future investigations.
Signaling from inside the ribosome
Most previous models assumed that binding of the SRP or other cellular machineries to the RNC occurs when signal sequences become exposed outside the ribosome. This view was initially challenged by the observation that the opening and closing of the Sec61p translocon is regulated by TMs in the nascent protein from inside the ribosome (212). More recently, multiple biochemical and crosslinking studies showed that, even when a signal sequence is still within the ribosome and has not emerged outside the exit tunnel, its presence enhances the binding of SRP to the RNC (38, 213) and helps recruit a regulatory protein RAMP4 to the Sec61p translocon (214). Further, in the GET pathway, the Bag6 complex is specifically recruited to the RNC when the C-terminal TM of the nascent protein emerges inside the ribosome (204). These results suggest that sequence or structural features of the nascent polypeptide inside the ribosome provide ‘signals’ that can be transmitted to the ribosome and lead to the recruitment of cellular factors. The nature of ribosome structural changes that underlie these signaling events, the mechanisms ensuring the specificity of these ‘signals’, and their precise roles in the respective cellular pathway are important questions for future studies.
SRP-dependent targeting to other translocons
Although SecYEG (or Sec61p) is a central protein conducting channel where many co- and post-translational pathways converge, membrane insertion of a subset of membrane proteins requires the translocase YidC, a member of the YidC/Oxa1/Alb3 family of proteins that facilitate the insertion and assembly of membrane proteins (see (215–217) for more comprehensive reviews). Although some of YidC’s function are carried out through cooperation with the SecYEG machinery (218), increasing evidence show that YidC can act independently of SecYEG to mediate the insertion of a number of proteins, including phage procoat proteins (219, 220), the mechanosensitive channel MscL (221, 222), and subunit c of the F1F0 ATP synthase (223, 224). In many studies, the targeting of MscL and the F1F0 subunits to YidC appears to be dependent on the cotranslational SRP/FtsY machinery (225–227). As noted earlier, the cpSRP targets LHC proteins to Alb3, the YidC homologue in chloroplasts. The structure (228, 229) and mechanism of YidC as an independent membrane protein insertase, how it interacts with the ribosome and the nascent polypeptide, and how it interfaces with the SRP targeting machinery remain to be determined. The decision-making process that allows the SRP to route a subset of its substrate proteins to the YidC instead of SecYEG translocon also remain to be elucidated, and will likely reveal additional layers of nuance and regulation in this pathway.
Translation-independent targeting of membrane proteins
Although targeted delivery of membrane proteins based on signals embedded in the nascent polypeptide has been long established, a recent study provided evidence for an alternative pathway(s) that localizes proteins to the target membrane in a translation-independent manner, based on cis-acting elements in the TM domain-encoding sequences of the mRNA (230). It was hypothesized that codons for hydrophobic amino acids in the TM domains are highly enriched in uracil content, which could provide a distinctive signature for these mRNAs to enable their recognition and targeted delivery to the membrane (231). The components, pathways and mechanisms of translation-independent targeting of membrane proteins and the contribution of these pathways to proper membrane protein localization within cells remain open questions that invite more investigations.
SUMMARY POINTS.
- SRP and SRP receptor catalyze the cotranslational delivery of membrane and secretory proteins to translocation machineries on the target membrane.
- Signal sequences allow nascent proteins to engage the correct cellular biogenesis machinery, and thus be directed to their proper cellular destination.
- SRP recognizes its cargos through bidentate interactions with the signal sequence and the ribosome. Likewise, the SRP receptor localizes to the target membrane through bidentate interactions with the phospholipid membrane and the SecYEG/Sec61p translocon.
- Two homologous GTPases in the SRP and SRP receptor use a unique GTPase cycle to drive and regulate the capture, delivery and unloading of cargo during protein targeting. They represent a growing class of dimerization-activated GTPases.
- The fidelity of substrate selection by the SRP is achieved through a combination of binding, induced fit, and kinetic proofreading mechanisms.
- The SRP RNA orchestrates global reorganization of the SRP, which enables rapid SRP–SRP receptor GTPase assembly in response to cargo binding.
- Eukaryotic SRP contains an additional Alu-domain that arrests translation elongation, which may provide a longer time window for the SRP in larger eukaryotic cells.
- Chloroplast SRP is dedicated to the delivery of fully synthesized LHC proteins, and has evolved unique molecular strategies to meet the challenges of post-translational membrane protein targeting.
FUTURE ISSUES.
- How is the translating ribosome productively handed over from the targeting to translocation machinery?
- What are the molecular codes that comprise the signal sequence?
- How are nascent proteins sorted among the myriad of protein biogenesis factors at the ribosome exit site and commit to the correct biogenesis pathway?
- Does a nascent polypeptide inside the ribosome tunnel ‘signal’ the ribosome to recruit specific factors, and how?
- How does the SRP route a subset of its substrates to the YidC or other membrane translocases instead of SecY/Sec61p?
- Does information embedded in the mRNA direct proteins to the membrane, and how?
Acknowledgments
We are indebted to Sandra Schmid, Jennifer Doudna, Peter Walter, Douglas Rees, Raymond Deshaies and Bil Clemons for support and insightful discussions over the years. S.S. was supported by NIH grant GM078024, and career awards from the Henry and Camille Dreyfus foundation, the Arnold and Mabel Beckman foundation, and the David and Lucile Packard foundation. D.A. was supported by NIH/NRSA training grant 5T32GM07616. X.Z. was supported by a fellowship from the Ulric B. and Evelyn L. Bray Endowment Fund.
Glossary
Protein targeting
the process of delivering newly synthesized protein to specific organelles in the cell
Cotranslational targeting
a mode of protein targeting in which the nascent protein is delivered while still attached to the translating ribosome
Post-translational targeting
a mode of protein targeting in which a fully synthesized nascent protein is delivered after release from the ribosome
Translocon
a protein complex that mediates the translocation or integration of proteins in the membrane bilayer. Used interchangeably with ‘translocation machinery’ and ‘translocase’
Signal sequence
transferable, cis-acting element on the nascent polypeptide that engages protein targeting machineries and mediates proper localization of the protein
P-loop GTPases
the most populous protein fold in nucleotide hydrolases, which uses the binding and hydrolysis of GTP to regulate cellular functions
Switch II loop
A structural segment in _Ras_-type signaling GTPases that interacts with effector proteins and moves upon GTP hydrolysis
Chromodomain
chromatin organization modifier domain, a highly conserved protein domain in eukaryotes often involved in chromatin remodeling
Ankyrin repeat
a 33-residue protein motif, which folds cooperatively with neighboring repeats and provide one of the most common protein interaction motifs
AAA+ disaggregases
a family of ATPases Associated with diverse cellular Activities that mediate ATP-dependent remodeling of protein aggregates
Acronyms
SRP
signal recognition particle
SR
signal recognition particle receptor
RNC
ribosome-nascent chain complex
Tat
the Twin-Arginine-Translocase system, comprised of TatA, TatB, and TatC proteins, which forms complexes that can transport folded proteins across the membrane
GET
the Guided Entry of Tail-anchor pathway, which mediates the post-translational targeting of tail-anchored membrane proteins to the ER membrane
GTPase
guanosine 5′-triphosphate (GTP) hydrolase
ATPase
adenosine 5′-triphosphate (ATP) hydrolase
Hsp70
the 70-kDa heat shock proteins, a family of ubiquitously expressed molecular chaperones that facilitate protein folding and biogenesis
NAC
nascent polypeptide associated complex, a heterodimeric complex which binds eukaryotic ribosomes in close proximity to the emerging nascent protein
LHCP
Light Harvesting Chlorophyll a,b-binding proteins, which form the antenna complex on photosynthetic centers in green plants
Literature Cited
- 1.Cross BCS, Sinning I, Luirink J, High S. Delivering proteins for export from the cytosol. Nature Rev Mol Cell Biol. 2009;10:255–64. doi: 10.1038/nrm2657. [DOI] [PubMed] [Google Scholar]
- 2.Driessen AJ, Nouwen N. Protein translocation across the bacterial cytoplasmic membrane. Annu Rev Biochem. 2008;77:643–67. doi: 10.1146/annurev.biochem.77.061606.160747. [DOI] [PubMed] [Google Scholar]
- 3.Pool MR. Signal recognition particles in chloroplasts, bacteria, yeast and mammals. Mole Membr Biol. 2005;22:3–15. doi: 10.1080/09687860400026348. [DOI] [PubMed] [Google Scholar]
- 4.Shao S, Hegde RS. Membrane protein insertion at the endoplasmic reticulum. Ann Rev Cell Dev Biol. 2011;27:25–56. doi: 10.1146/annurev-cellbio-092910-154125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilmore R, Blobel G, Walter P. Protein translocation across the endoplasmic reticulum: 1. Detection in the microsomal membrane of a receptor for the signal recognition particle. J Cell Biol. 1982a;95:463–9. doi: 10.1083/jcb.95.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilmore R, Walter P, Blobel G. Protein translocation across the endoplasmic reticulum. II Isolation and characterization of the signal recognition particle receptor. J Cell Biol. 1982b;95:470–7. doi: 10.1083/jcb.95.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walter P, Blobel G. Purification of a membrane-associated protein complex required for protein translocation across the endoplasmic reticulum. Proc Natl Acad Sci U S A. 1980;77:7112–6. doi: 10.1073/pnas.77.12.7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walter P, Blobel G. Disassembly and reconstitution of signal recognition particle. Cell. 1983;34:525–33. doi: 10.1016/0092-8674(83)90385-9. [DOI] [PubMed] [Google Scholar]
- 9.Walter P, Gilmore R, Blobel G. Protein translocation across the endoplasmic reticulum. Cell. 1984;38:5–8. doi: 10.1016/0092-8674(84)90520-8. [DOI] [PubMed] [Google Scholar]
- 10.Bernstein HD, Poritz MA, Strub K, Hoben PJ, Brenner S, Walter P. Model for signal sequence recognition from amino-acid sequence of 54K subunit of signal recognition particle. Nature. 1989;340:482–6. doi: 10.1038/340482a0. [DOI] [PubMed] [Google Scholar]
- 11.Bernstein HD, Zopf D, Freymann DM, Walter P. Functional substitution of the signal recognition particle 54-kDa subunit by its Escheirchia coli homolog. Proc Natl Acad Sci U S A. 1993;90:5229–34. doi: 10.1073/pnas.90.11.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Römisch K, Webb J, Herz J, Prehn S, Frank R, et al. Homology of the 54K protein of signal recognition particle, docking protein, and two E. coli proteins with putative GTP-binding domains. Nature. 1989;340:478–82. doi: 10.1038/340478a0. [DOI] [PubMed] [Google Scholar]
- 13.Powers T, Walter P. Co-translational protein targeting catalyzed by the Escherichia coli signal recognition particle and its receptor. EMBO J. 1997;16:4880–6. doi: 10.1093/emboj/16.16.4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batey RT, Rambo RP, Lucast L, Rha B, Doudna JA. Crystal structure of the ribonucleoprotein core of the signal recognition particle. Science. 2000;287:1232–9. doi: 10.1126/science.287.5456.1232. [DOI] [PubMed] [Google Scholar]
- 15.Janda CY, Li J, Oubridge C, Hernandez H, Robinson CV, Nagai K. Recognition of a signal peptide by the signal recognition particle. Nature. 2010;465:507–10. doi: 10.1038/nature08870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keenan RJ, Freymann DM, Walter P, Stroud RM. Crystal structure of the signal sequence binding subunit of the signal recognition particle. Cell. 1998;94:181–91. doi: 10.1016/s0092-8674(00)81418-x. [DOI] [PubMed] [Google Scholar]
- 17.Egea PF, Shan S, Napetschnig J, Savage DF, Walter P, Stroud RM. Substrate twinning activates the signal recognition particle and its receptor. Nature. 2004;427:215–21. doi: 10.1038/nature02250. [DOI] [PubMed] [Google Scholar]
- 18.Focia PJ, Shepotinovskaya IV, Seidler JA, Freymann DM. Heterodimeric GTPase Core of the SRP Targeting Complex. Science. 2004;303:373–7. doi: 10.1126/science.1090827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parlitz R, Eitan A, Stjepanovic G, Bahari L, Bange G, Bibi E, Sinning I. Escherichia coli signal recognition particle receptor FtsY contains an essential and autonomous membrane-binding amphipathic helix. J Biol Chem. 2007;282:32176–84. doi: 10.1074/jbc.M705430200. [DOI] [PubMed] [Google Scholar]
- 20.Weiche B, Burk J, Angelini S, Schiltz E, Thumfart JO, Koch HG. A cleavable N-terminal membrane anchor is involved in membrane binding of the Escherichia coli SRP receptor. J Mol Biol. 2008;377:761–73. doi: 10.1016/j.jmb.2008.01.040. [DOI] [PubMed] [Google Scholar]
- 21.Goldshmidt H, Sheiner L, Butikofer P, Roditi I, Uliel S, Gunzel M, Engstler M, Michaeli S. Role of protein translocation pathways across the endoplasmic reticulum in Trypanosoma brucei. J Biol Chem. 2008;283:32085–98. doi: 10.1074/jbc.M801499200. [DOI] [PubMed] [Google Scholar]
- 22.Muller L, de Escauriaza MD, Lajoie P, Theis M, Jung M, Muller A, Burgard C, Greiner M, Snapp EL, Dudek J, Zimmermann R. Evolutionary gain of function for the ER membrane protein Sec62 from yeast to humans. Mol Biol Cell. 2010;21:691–703. doi: 10.1091/mbc.E09-08-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deshaies RJ, Koch BD, Werner-Washburne M, Craig EA, Schekman R. A subfamily of stress proteins faciliates translocation of secretory and mitochondrial precursor polypeptides. Nature. 1988;332:800–5. doi: 10.1038/332800a0. [DOI] [PubMed] [Google Scholar]
- 24.Frobel J, Rose P, Muller M. Twin-arginine-dependent translocation of folded proteins. Philos Trans R Soc Lond B Biol Sci. 2012;367:1029–46. doi: 10.1098/rstb.2011.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hegde RS, Keenan RJ. Tail-anchored membrane protein insertion into the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2011;12:787–98. doi: 10.1038/nrm3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Natale P, Bruser T, Driessen AJ. Sec- and Tat-mediated protein secretion across the bacterial cytoplasmic membrane – distinct translocases and mechanisms. Biochim Biophys Acta. 2008;1778:1735–56. doi: 10.1016/j.bbamem.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 27.Gierasch LM. Signal sequences. Biochemistry. 1989;28:923–30. doi: 10.1021/bi00429a001. [DOI] [PubMed] [Google Scholar]
- 28.von Heijne G. Signal sequences: The limits of variation. J Mol Biol. 1985;184:99–105. doi: 10.1016/0022-2836(85)90046-4. [DOI] [PubMed] [Google Scholar]
- 29.Krieg UC, Walter P, Johnson AE. Photocrosslinking of the signal sequence of nascent preprolactin to the 54-kilodalton polypeptide of the signal recognition particle. Proc Natl Acad Sci U S A. 1986;83:8604–8. doi: 10.1073/pnas.83.22.8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurzchalia TV, Wiedmann M, Girshovich AS, Bochkareva ES, Bielka H, Rapoport TA. The signal sequence of nascent preprolactin interacts with the 54K polypeptide of the signal recognition particle. Nature. 1986;320:634–6. doi: 10.1038/320634a0. [DOI] [PubMed] [Google Scholar]
- 31.Zopf D, Bernstein HD, Johnson AE, Walter P. The methionine-rich domain of the 54 kd protein subunit of the signal recognition particle contains an RNA binding site and can be crosslinked to a signal sequence. EMBO J. 1990;9:4511–7. doi: 10.1002/j.1460-2075.1990.tb07902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hainzl T, Huang S, Merilainen G, Brannstrom K, Sauer-Eriksson AE. Structural basis of signal sequence recognition by the signal recognition particle. Nature Struct Molec Biol. 2011;18:389–91. doi: 10.1038/nsmb.1994. [DOI] [PubMed] [Google Scholar]
- 33.Hainzl T, Huang S, Sauer-Eriksson AE. Interaction of signal-recognition particle 54 GTPase domain and signal recognition particle RNA in the free signal-recognition particle. Proc Natl Acad Sci. 2007;104:14911–6. doi: 10.1073/pnas.0702467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosendal KR, Wild K, Montoya G, Sinning I. Crystal structure of the complete core of archaeal signal recognition paricle and implications for interdomain communication. Proc Natl Acad Sci. 2003;100:14701–6. doi: 10.1073/pnas.2436132100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bradshaw N, Walter P. The Signal Recognition Particle (SRP) RNA links conformational changes in the SRP to protein targeting. Mol Biol Cell. 2007;18:2728–34. doi: 10.1091/mbc.E07-02-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bradshaw N, Neher SB, Booth DS, Walter P. Signal sequences activate the catalytic switch of SRP RNA. Science. 2009;323:127–30. doi: 10.1126/science.1165971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swain JF, Gierasch LM. Signal peptides bind and aggregate RNA: an alternative explanation for GTPase inhibition in the signal recognition particle. J Biol Chem. 2001;276:12222–7. doi: 10.1074/jbc.M011128200. [DOI] [PubMed] [Google Scholar]
- 38.Bornemann T, Jockel J, Rodnina MV, Wintermeyer W. Signal sequence-independent membrane targeting of ribosomes containing short nascent peptides within the exit tunnel. Nat Struct Mol Biol. 2008;15:494–9. doi: 10.1038/nsmb.1402. [DOI] [PubMed] [Google Scholar]
- 39.Flanagan JJ, Chen JC, Miao Y, Shao Y, Lin J, Bock PE, Johnson AE. Signal recognition particle binds to ribosome-bound signal sequences with fluorescence-detected subnanomolar affinity that does not diminish as the nascent chain lengthens. J Biol Chem. 2003;278:18628–37. doi: 10.1074/jbc.M300173200. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, Rashid R, Wang K, Shan S. Sequential checkpoints govern fidelity during co-translational protein targeting. Science. 2010;328:757–60. doi: 10.1126/science.1186743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gu SQ, Peske F, Wieden HJ, Rodnina MV, Wintermeyer W. The signal recognition particle binds to protein L23 at the peptide exit of the Escherichia coli ribosome. RNA. 2003;9:566–73. doi: 10.1261/rna.2196403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pool MR, Stumm J, Fulga TA, Sinning I, Dobberstein B. Distinct modes of signal recognition particle interaction with the ribosome. Science. 2002;297:1345–8. doi: 10.1126/science.1072366. [DOI] [PubMed] [Google Scholar]
- 43.Halic M, Becker T, Pool MR, Spahn CMT, Grassucci RA, Frank J, Beckmann R. Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature. 2004;427:808–14. doi: 10.1038/nature02342. [DOI] [PubMed] [Google Scholar]
- 44.Halic M, Blau M, Becker T, Mielke T, Pool MR, Wild K, Sinning I, Beckmann R. Following the signal sequence from ribosomal tunnel exit to signal recognition particle. Nature. 2006;444:507–11. doi: 10.1038/nature05326. [DOI] [PubMed] [Google Scholar]
- 45.Schaffitzel C, Oswald M, Berger I, Ishikawa T, Abrahams JP, Koerten HK, Koning RI, Ban N. Structure of the E. coli signal recognition particle bound to a translating ribosome. Nature. 2006;444:503–6. doi: 10.1038/nature05182. [DOI] [PubMed] [Google Scholar]
- 46.Saraogi I, Zhang D, Chandrasekaran S, Shan S. Site-specific fluorescent labeling of nascent proteins on the translating ribosome. J Am Chem Soc. 2011;133:14936–9. doi: 10.1021/ja206626g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mircheva M, Boy D, Weiche B, Hucke F, Graumann P, Koch H-G. Predominant membrane localization is an essential feature of the bacterial signal recognition particle receptor. BMC Biology. 2009;7 doi: 10.1186/1741-7007-7-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rubio A, Jiang X, Pogliano K. Localization of translocation complex components in Bacillus subtilis: enrichment of the signal recognition particle receptor at early sporulation septa. J Bacteriol. 2005;187:5000–2. doi: 10.1128/JB.187.14.5000-5002.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luirink J, Hagen-Jongman C, ten Weijden CM, Oudega B, High S, Dobberstein B, Kusters R. An alternative protein targeting pathway in Escherichia coli: studies on the role of FtsY. EMBO J. 1994;13:2289–96. doi: 10.1002/j.1460-2075.1994.tb06511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Leeuw E, te Kaat K, Moser C, Memestrina G, Demel R, de Kruijff B, Oudegam B, Luirink J, Sinning I. Anionic phospholipids are involved in membrane association of FtsY and stimulate its GTPase activity. EMBO J. 2000;19:531–41. doi: 10.1093/emboj/19.4.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lam VQ, Akopian D, Rome M, Shen Y, Henningsen D, Shan S. Lipid activation of the SRP receptor provides spatial coordination of protein targeting. J Cell Biol. 2010;190:623–35. doi: 10.1083/jcb.201004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bahari L, Parlitz R, Eitan A, Stjepanovic G, Bochkareva ES, Sining I, Bibi E. Membrane targeting of ribosomes and their release require distinct and separable functions of FtsY. J Biol Chem. 2007;282:32168–75. doi: 10.1074/jbc.M705429200. [DOI] [PubMed] [Google Scholar]
- 53.Eitan A, Bibi E. The core Escherichia coli signal recognition particle receptor contains only the N and G domains of FtsY. J Bacteriol. 2004;186:2492–4. doi: 10.1128/JB.186.8.2492-2494.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marty N, Rajalingam D, Kight AD, Lewis N, Fologea D, Kumar TKS, Henry R, Goforth RL. The membrane binding motif of chloroplast signal recognition particle receptor (cpFtsY) regulates GTPase activity. J Biol Chem. 2009;284:14891–903. doi: 10.1074/jbc.M900775200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Montoya G, Svensson C, Luirink J, Sinning I. Crystal structure of the NG domain from the signal recognition particle receptor FtsY. Nature. 1997;385:365–8. doi: 10.1038/385365a0. [DOI] [PubMed] [Google Scholar]
- 56.Erez E, Stjepanovic G, Zelazny AM, Brugger B, Sinning I, Bibi E. Genetic evidence for functional interaction of the Escherichia coli signal recognition particle receptor with acidic lipids in vivo. J Biol Chem. 2010;285:40508–14. doi: 10.1074/jbc.M110.140921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gold VAM, Robson A, Bao H, Romantsov T, Duong F, Collinson I. The action of cardiolipin on the bacterial translocon. Proc Natl Acad Sci. 2010 doi: 10.1073/pnas.0914680107. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hendrick JP, Wickner W. SecA protein needs both acidic phospholipids and secY/E protein for functional high affinity binding to the Escherichia coli plasma membrane. J Biol Chem. 1991;266:24596–600. [PubMed] [Google Scholar]
- 59.Lill R, Dowhan W, Wickner W. The ATPase activity of secA is regulated by acidic phospholipids, secY, and the leader and mature domains of precursor proteins. Cell. 1990;60:271–80. doi: 10.1016/0092-8674(90)90742-w. [DOI] [PubMed] [Google Scholar]
- 60.de Vruje T, de Swart RL, Dowhan W, Tommassen J, de Kruijff B. Phosphotidylglycerol is involved in protein translocation across Escherichia coli inner membranes. Nature. 1988;334:173–5. doi: 10.1038/334173a0. [DOI] [PubMed] [Google Scholar]
- 61.Kusters R, Dowhan W, de Kruijff B. Negatively charged phospholipids restore prePhoE translocation across phosphatidylglycerol-depleted Escherichia coli inner membranes. J Biol Chem. 1991;266:8659–62. [PubMed] [Google Scholar]
- 62.Ridder ANJA, Kuhn A, Killian JA, de Kruijff B. Anionic lipids stimulate Sec-independent insertion of a membrane protein lacking charged amino acid side chains. EMBO Rep. 2001;2:403–8. doi: 10.1093/embo-reports/kve087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Angelini S, Deitermann S, Koch HG. FtsY, the bacterial signal recogntion particle receptor, interacts functionally and physically with the secYEG translocon. EMBO Rep. 2005;6:476–81. doi: 10.1038/sj.embor.7400385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Angelini S, Boy D, Schiltz E, Koch HG. Membrane binding of the bacterial signal recognition particle receptor involves two distinct binding modes. J Cell Biol. 2006;174:715–24. doi: 10.1083/jcb.200606093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kuhn P, Weiche B, Sturm L, Sommer E, Drepper F, Warscheid B, Sourjik V, Koch HG. The bacterial SRP receptor, SecA and the ribosome use overlapping binding sites on the SecY translocon. Traffic. 2011;12:563–78. doi: 10.1111/j.1600-0854.2011.01167.x. [DOI] [PubMed] [Google Scholar]
- 66.Tajima S, Lauffer L, Rath VL, Walter P. The signal recognition particle receptor is a complex that contains two distinct polypeptide chains. J Cell Biol. 1986;103:1167–78. doi: 10.1083/jcb.103.4.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schwartz T, Blobel G. Structural basis for the function of the b-subunit of the eukaryotic signal recognition particle receptor. Cell. 2003;112:793–803. doi: 10.1016/s0092-8674(03)00161-2. [DOI] [PubMed] [Google Scholar]
- 68.Miller JD, Tajima S, Lauffer L, Walter P. The β subunit of the signal recognition particle receptor is a transmembrane GTPase that anchors the a subunit, a peripheral membrane GTPase, to the endoplasmic reticulum. J Cell Biol. 1995;128:273–82. doi: 10.1083/jcb.128.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Helmers J, Schmidt D, Glavy JS, Blobel G, Schwartz T. The b-subunit of the protein-conducting channel of the endoplasmic reticulum functions as the guanine nucleotide exchange factor for the b-subunit of the signal recognition particle receptor. J Biol Chem. 2003;278:23686–90. doi: 10.1074/jbc.C300180200. [DOI] [PubMed] [Google Scholar]
- 70.Wittke S, Dunnwald M, Albertsen M, Johnsson N. Recognition of a subset of signal sequences by Ssh1p, a Sec61p-related protein in the membrane of endoplasmic reticulum of yeast Saccharomyces cerevisiae. Mol Biol Cell. 2002;13:2223–32. doi: 10.1091/mbc.01-10-0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jiang Y, Cheng Z, Mandon EC, Gilmore R. An interaction between the SRP receptor and the translocon is critical during cotranslational protein translocation. J Cell Biol. 2003;180:1149–61. doi: 10.1083/jcb.200707196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Freymann DM, Keenan RJ, Stroud RM, Walter P. Structure of the conserved GTPase domain of the signal recognition particle. Nature. 1997;385:361–4. doi: 10.1038/385361a0. [DOI] [PubMed] [Google Scholar]
- 73.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature. 1990;348:125–8. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- 74.Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–49. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 75.Gasper R, Meyer S, Gotthardt K, Sirajuddin M, Wittinghofer A. It takes two to tango: regulation of G proteins by dimerization. Nature Rev Mol Cell Biol. 2009;10:423–9. doi: 10.1038/nrm2689. [DOI] [PubMed] [Google Scholar]
- 76.Bange G, Kummerer N, Grudnik P, Lindner R, Petzold G, Kressler D, Hurt E, Wild K, Sinning I. Structural basis for the molecular evolution of SRP-GTPase activation by protein. Nature Struct Molec Biol. 2011;18:1376–80. doi: 10.1038/nsmb.2141. [DOI] [PubMed] [Google Scholar]
- 77.Chappie JS, Acharya S, Leonard M, Schmid SL, Dyda F. G domain dimerization controls dynamin’s assembly-stimulated GTPase activity. Nature. 2010;465:435–40. doi: 10.1038/nature09032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leipe DD, Wolf YI, Koonin EV, Aravid L. Classificaiton and evolution of P-loop GTPases and related ATPases. J Mol Biol. 2002;317:41–72. doi: 10.1006/jmbi.2001.5378. [DOI] [PubMed] [Google Scholar]
- 79.Freymann DM, Keenan RJ, Stroud RM, Walter P. Functional changes in the structure of the SRP GTPase on binding GDP and Mg2+GDP. Nat Struct Biol. 1999;6:793–801. doi: 10.1038/11572. [DOI] [PubMed] [Google Scholar]
- 80.Gawronski-Salerno J, Coon YJS, Focia PJ, Freymann DM. X-ray structure of the T. Aquaticus Ftsy:GDP complex suggests functional roles for the C-terminal helix of the SRP GTPases. Proteins. 2006;66:984–95. doi: 10.1002/prot.21200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Padmanabhan W, Freymann DM. The conformation of bound GMPPNP suggests a mechanism for gating the active site of the SRP GTPase site. Structure. 2001;9:859–63. doi: 10.1016/s0969-2126(01)00641-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Reyes CL, Rutenber E, Walter P, Stroud RM. X-ray structures of the signal recognition particle receptor reveal targeting cycle intermediates. PloS ONE. 2007;2:e607. doi: 10.1371/journal.pone.0000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peluso P, Shan S, Nock S, Herschlag D, Walter P. Role of SRP RNA in the GTPase cycles of Ffh and FtsY. Biochemistry. 2001;40:15224–33. doi: 10.1021/bi011639y. [DOI] [PubMed] [Google Scholar]
- 84.Shan S, Schmid SL, Zhang X. Signal recognition particle (SRP) and SRP receptor: a new paradigm for multi-state regulatory GTPases. Biochemistry. 2009;48:6696–704. doi: 10.1021/bi9006989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang X, Kung S, Shan S. Demonstration of a two-step mechanism for assembly of the SRP-SRP receptor complex: implications for the catalytic role of SRP RNA. J Mol Biol. 2008;381:581–93. doi: 10.1016/j.jmb.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Estrozi LF, Boehringer D, Shan S, Ban N, Schaffitzel C. Cryo-EM structure of the E. coli translating ribosome in complex with SRP and its receptor. Nat Struct Mol Biol. 2011;18:88–90. doi: 10.1038/nsmb.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang X, Lam VQ, Mou Y, Kimura T, Chung J, Chandrasekar S, Winkler J, Mayo S, Shan S. Direct visualization reveals dynamics of a transient intermediate during proten assembly. Proc Natl Acad Sci U S A. 2011;108:6450–5. doi: 10.1073/pnas.1019051108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shan S, Walter P. Induced Nucleotide Specificity in a GTPase. Proc Natl Acad Sci USA. 2003;100:4480–5. doi: 10.1073/pnas.0737693100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shan S, Stroud R, Walter P. Mechanism of association and reciprocal activation of two GTPases. Plos Biology. 2004;2:e320. doi: 10.1371/journal.pbio.0020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gawronski-Salerno J, Freymann DM. Structure of the GMPPNP-stabilized NG domain complex of the SRP GTPases Ffh and FtsY. J Struct Biol. 2007;158:122–8. doi: 10.1016/j.jsb.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Neher SB, Bradshaw N, Floor SN, Gross JD, Walter P. SRP RNA controls a conformational switch regulating the SRP-SRP receptor interaction. Nat Struct Mol Biol. 2008;15:916–23. doi: 10.1038/nsmb.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shepotinovskaya IV, Freymann DM. Conformational change of the N-domain on formation of the complex between the GTPase domains of Thermus aquaticus Ffh and FtsY. Biochemica et Biophysica Acta. 2001;1597:107–14. doi: 10.1016/s0167-4838(02)00287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Connolly T, Rapiejko PJ, Gilmore R. Requirement of GTP hydrolysis for dissociation of the signal recognition particle from its receptor. Science. 1991;252:1171–3. doi: 10.1126/science.252.5009.1171. [DOI] [PubMed] [Google Scholar]
- 94.Peluso P, Herschlag D, Nock S, Freymann DM, Johnson AE, Walter P. Role of 4.5S RNA in assembly of the bacterial signal recognition particle with its receptor. Science. 2000;288:1640–3. doi: 10.1126/science.288.5471.1640. [DOI] [PubMed] [Google Scholar]
- 95.Zhang X, Schaffitzel C, Ban N, Shan S. Multiple conformational changes in a GTPase complex regulate protein targeting. Proc Natl Acad Sci. 2009;106:1754–9. doi: 10.1073/pnas.0808573106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Braig D, Mircheva M, Sachelaru I, van der Sluis EO, Sturn L, Beckmann R, Koch HG. Signal sequence-independent SRP-SR complex formation at the membrane suggests an alternative targeting pathway within the SRP cycle. Mol Biol Cell. 2011;22:2309–23. doi: 10.1091/mbc.E11-02-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stjepanovic G, Kapp K, Bange G, Graf C, Parlitz R, Wild K, Mayer MP, Sinning I. Lipids trigger a conformational switch that regulates signal recognition particle (SRP0-mediated protein targeting. J Biol Chem. 2011;286:23489–97. doi: 10.1074/jbc.M110.212340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shan S, Chandrasekar S, Walter P. Conformational changes in the GTPase modules of SRP and its receptor drive initiation of protein translocation. J Cell Biol. 2007;178:611–20. doi: 10.1083/jcb.200702018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Halic M, Gartmann M, Schlenker O, Mielke T, Pool MR, Sinning I, Beckmann R. Signal recognition particle receptor exposes the ribosomal translocon binding site. Science. 2006;312:745–7. doi: 10.1126/science.1124864. [DOI] [PubMed] [Google Scholar]
- 100.Kaiser CA, Preuss D, Grisafi P, Botstein D. Many random sequences functionally replace the secretion signal sequence of yeast invertase. Science. 1987;235:312–7. doi: 10.1126/science.3541205. [DOI] [PubMed] [Google Scholar]
- 101.Zheng N, Gierasch LM. Signal sequences: the same yet different. Cell. 1996;86:849–52. doi: 10.1016/s0092-8674(00)80159-2. [DOI] [PubMed] [Google Scholar]
- 102.Jungnickel B, Rapoport TA. A posttargeting signal sequence recognition event in the endoplasmic reticulum membrane. Cell. 1995;82:261–70. doi: 10.1016/0092-8674(95)90313-5. [DOI] [PubMed] [Google Scholar]
- 103.Kundel TA, Bebenek K. DNA replication fidelity. Ann Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 104.Uptain SM, Kane CM, Chamberlin MJ. Basic mechanisms of transcript elongation and its regulation. Ann Rev Biochem. 1997;66:117–72. doi: 10.1146/annurev.biochem.66.1.117. [DOI] [PubMed] [Google Scholar]
- 105.Semlow DR, Staley JP. Staying on message: ensuring fidelity in pre-mRNA splicing. TIBS. 2012;37:263–73. doi: 10.1016/j.tibs.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fersht AR, Kaethner MM. Enzyme hyperspecificity. Rejection of threonine by the valyl-tRNA synthetase by misacylation and hydrolytic editing. Biochemistry. 1976;15:3342–6. doi: 10.1021/bi00660a026. [DOI] [PubMed] [Google Scholar]
- 107.Rodnina MV, Wintermeyer W. Ribosome fidelity: tRNA discrimination, proofreading and induced fit. TIBS. 2001;26:124–30. doi: 10.1016/s0968-0004(00)01737-0. [DOI] [PubMed] [Google Scholar]
- 108.Rosenblad MA, Gorodkin J, Knudsen B, Zwieb C, Samuelsson T. SRPDB: Signal Recongnition Particle Database. Nuc Acids Res. 2003;31:363–4. doi: 10.1093/nar/gkg107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Poritz MA, Strub K, Walter P. Human SRP RNA and E. coli. 4.5S RNA contain a highly homologous structural domain. Cell. 1988;55:4–6. doi: 10.1016/0092-8674(88)90003-7. [DOI] [PubMed] [Google Scholar]
- 110.Batey RT, Sagar MB, Doudna JA. Structural and energetic analysis of RNA recognition by a universally conserved protein from the signal recognition particle. J Mol Biol. 2001;307:229–46. doi: 10.1006/jmbi.2000.4454. [DOI] [PubMed] [Google Scholar]
- 111.Buskiewicz I, Kubarenko A, Peske F, Rodnina MV, Wintermeyer W. Domain rearrangement of SRP protein Ffh upon binding 4.5S RNA and the SRP receptor FtsY. RNA. 2005;11:947–57. doi: 10.1261/rna.7242305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Buskiewicz I, Peske F, Wieden HJ, Gryczynski I, Rodnina MV, Wintermeyer W. Conformations of the signal recognition particle protein Ffh from Escherichia coli as determined by FRET. J Mol Biol. 2005;351:417–30. doi: 10.1016/j.jmb.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 113.Mainprize IL, BEniac DR, Falkovskala E, Cleverley RM, Gierasch LM, Ottensmeyer FP, Andrews DW. The structure of Escherichia coli signal recognition particle revealed by scanning transmission electron microscopy. Mol Biol Cell. 2006;17:5063–74. doi: 10.1091/mbc.E06-05-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Buskiewicz I, Jockel J, Rodnina MV, Wintermeyer W. Conformation of the signal recognition particle in ribosomal targeting complexes. RNA. 2009;15:44–54. doi: 10.1261/rna.1285609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jagath JR, Matassova NB, de Leeuw E, Warnecke JM, Lentzen G, Rodnina MV, Luirink J, Wintermeyer W. Important role of the tetraloop region of 4.5S RNA in SRP binding to its receptor FtsY. RNA. 2001;7:293–301. doi: 10.1017/s1355838201002205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Siu F-Y, Spanggord RJ, Doudna JA. SRP RNA provides the physiologically essential GTPase activation function in cotranslational protein targeting. RNA. 2006;13:1–11. doi: 10.1261/rna.135407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shen K, Shan S. A transient tether between the SRP RNA and SRP receptor ensures efficient cargo delivery during cotranslational protein targeting. Proc Natl Acad Sci U S A. 2010;107:7698–703. doi: 10.1073/pnas.1002968107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Spanggord RJ, Siu F, Ke A, Doudna JA. RNA-mediated interaction between the peptide-binding and GTPase domains of the signal recognition particle. Nature Struct Molec Biol. 2005;12:1116–22. doi: 10.1038/nsmb1025. [DOI] [PubMed] [Google Scholar]
- 119.Shen K, Zhang X, Shan S. Synergiestic action between the SRP RNA and translating ribosome allows efficient delivery of correct cargos during co-translational protein targeting. RNA. 2011;17:892–902. doi: 10.1261/rna.2610411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Attaide SF, Schmitz N, Shenk K, Ke A, Shan S, Doudna A, Ban N. The crystal structure of the Signal Recognition Particle in complex with its receptor. Science. 2011;381:881–6. doi: 10.1126/science.1196473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.de Plessis DJ, Nouwen N, Diriessen AJ. The Sec translocase. Biochim Biophys Acta. 2011;1808:851–65. doi: 10.1016/j.bbamem.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 122.Johnson AE, van Waes MA. The translocon: A dynamic gateway at the ER membrane. Annu Rev Cell Dev Biol. 1999;18:799–842. doi: 10.1146/annurev.cellbio.15.1.799. [DOI] [PubMed] [Google Scholar]
- 123.Rapaport TA, Jungnickel B, Kutay U. Protein transport across the eukaryotic endoplasmic reticulum and bacterial inner membranes. Annu Rev Biochem. 1996;65:271–303. doi: 10.1146/annurev.bi.65.070196.001415. [DOI] [PubMed] [Google Scholar]
- 124.van der Berg B, Clemons WM, Collinson I, Modis Y, Hartmann E, Harrison SC, Rapoport TA. X-ray structure of a protein-conducting channel. Nature. 2003;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- 125.Cannon KS, Or E, Clemons WM, Jr, Shibata Y, Rapoport TA. Disulfide bridge formation between SecY and a translocating polypeptide localizes the translocation pore to the center of SecY. J Cell Biol. 2005;169:219–25. doi: 10.1083/jcb.200412019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Heinrich SU, Mothes W, Brunner H, Rapoport TA. The Sec61p complex mediates the integration of a membrane protein by alllowing lipid partitioning of the transmembrane domain. Cell. 2000;102:233–44. doi: 10.1016/s0092-8674(00)00028-3. [DOI] [PubMed] [Google Scholar]
- 127.Plath K, Mothes W, Wikinson BM, Sterling CJ, Rapoport TA. Signal sequence recognition in post-translational protein transport across the yeast ER membrane. Cell. 1998;94:795–807. doi: 10.1016/s0092-8674(00)81738-9. [DOI] [PubMed] [Google Scholar]
- 128.Becker T, Bhushan S, Jarasch A, Armache J-P, Funes S, Jossinet F, Gumbart J, Mielke T, Berninhausen O, Schulten K, Westhof E, Gilmore R, Mandon E, Beckmann R. Structure of Monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science. 2009;326:1367–73. doi: 10.1126/science.1178535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Beckmann R, Spahn CMT, Eswar N, Helmers J, Penczek PA, Sali A, Frank J, Blobel G. Architecture of the protein-conducting channel associated with the translating 80S ribosome. Cell. 2001;107:361–72. doi: 10.1016/s0092-8674(01)00541-4. [DOI] [PubMed] [Google Scholar]
- 130.Fraunfeld J, Gumbart J, Sluis EO, Funes S, Gartmann M, Beatrix B, Mielke T, Berninghausen O, Becker T, Schulten K, Beckmann R. Cryo-EM structure of the ribosome-SecYE complex in the membrane environment. Nature Struct Molec Biol. 2011;18:614–21. doi: 10.1038/nsmb.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.MItra K, Schaffitzel C, Shaikh T, Tanna F, Jenni S, Brooks CL, Ban N, Frank J. Structure of the E. coli protein conducting channel bound to a translating ribosome. Nature. 2005;438:318–24. doi: 10.1038/nature04133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cheng Z, Jiang Y, Mandon EC, Gilmore R. Identification of cytoplasmic residues of Sec61p involved in ribosome binding and cotranslational translocation. J Cell Biol. 2005;168:67–77. doi: 10.1083/jcb.200408188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Menetret JF, Schaletzky J, Clemons WM, Jr, Osborne AR, Skanland SS, Denison C, Gygi SP, Kirkpatrick DS, Park E, Ludtke SJ, Rapoport TA, Akey CW. Ribosome binding of a single copy of the SecY complex: implications for protein translocation. Mol Cell. 2007;28:1083–92. doi: 10.1016/j.molcel.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 134.Maity TS, Leonard CW, Rose MA, Fried HM, Weeks KM. Compartmentalization directs assembly of the signal recognition particle. Biochemistry. 2006;45:14955–64. doi: 10.1021/bi060890g. [DOI] [PubMed] [Google Scholar]
- 135.Maity TS, Weeks KM. A threefold RNA-protein interface in the signal recognition particle gates native complex assembly. J Mol Biol. 2007;369:512–24. doi: 10.1016/j.jmb.2007.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ciufo LF, Brown JD. Nuclear export of yeast signal recognition particle lacking Srp54p via the Xpo1p/Crm1p, NES-dependent pathway. Curr Biol. 2000;10:1256–64. doi: 10.1016/s0960-9822(00)00743-0. [DOI] [PubMed] [Google Scholar]
- 137.Grosshans H, Deinert K, Hurt E, Simos G. Biogenesis of the signal recognition particle (SRP) involves import of SRP proteins into the nucleolus, assembly with the SRP-RNA, and Xpo1p-mediated export. J Cell Biol. 2001;153:745–62. doi: 10.1083/jcb.153.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jacobson MR, Pederson T. Localization of signal recognition particle RNA in the nucleolus of mammalian cells. Proc Natl Acad Sci U S A. 1998;95:7981–6. doi: 10.1073/pnas.95.14.7981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Leung E, Brown JD. Biogenesis of the signal recognition particle. Biochem Soc Trans. 2010;38:1093–8. doi: 10.1042/BST0381093. [DOI] [PubMed] [Google Scholar]
- 140.Siegel V, Walter P. Elongation arrest is not a prerequisite for secretory protein translocation across the microsomal membrane. J Cell Biol. 1985;100:1913–21. doi: 10.1083/jcb.100.6.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Egea PF, Napetschnig J, Walter P, Stroud RM. Structures of SRP54 and SRP19, the two proteins that organize the ribonucleic core of the signal recognition particle. PloS ONE. 2008;3:e3528. doi: 10.1371/journal.pone.0003528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hainzl T, Huang S, Sauer-Eriksson AE. Structure of the SRP19-RNA complex and implication for signal recognition particle assembly. Nature. 2002;417:767–71. doi: 10.1038/nature00768. [DOI] [PubMed] [Google Scholar]
- 143.Hainzl T, Huang S, Sauer-Eriksson AE. Structural insights into SRP RNA: an induced-fit mechanism for SRP assembly. RNA. 2005;11:1043–50. doi: 10.1261/rna.2080205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Menichelli E, Isel C, Oubridge C, Nagai K. Protein-induced conformational changes of RNA during the assembly of human signal recognition particle. J Mol Biol. 2007;367:187–203. doi: 10.1016/j.jmb.2006.12.056. [DOI] [PubMed] [Google Scholar]
- 145.Oubridge C, Kuglstatter A, Jovine L, Nagai K. Crystal structure of SRP19 in complex with the S domain of SRP RNA and its implication for the assembly of the signal recognition particle. Molecular Cell. 2002;9:1251–61. doi: 10.1016/s1097-2765(02)00530-0. [DOI] [PubMed] [Google Scholar]
- 146.Wild K, Sinning I, Cusack S. Crystal structure of an early protein-RNA assembly complex of the signal recognition particle. Science. 2001;294:598–601. doi: 10.1126/science.1063839. [DOI] [PubMed] [Google Scholar]
- 147.Sauer-Eriksson AE, Hainzl T. S-domain assembly of the signal recognition particle. Curr Opin Struct Biol. 2003;13:64–70. doi: 10.1016/s0959-440x(02)00010-6. [DOI] [PubMed] [Google Scholar]
- 148.Iakhiaeva E, Yin J, Zwieb C. Identification of an RNA-binding domain in human SRP72. J Mol Biol. 2005;345:659–66. doi: 10.1016/j.jmb.2004.10.087. [DOI] [PubMed] [Google Scholar]
- 149.Iakhiaeva E, Bhuiyan SH, Yin J, Zwieb C. Protein SRP68 of human signal recognition particle: identification of the RNA and SRP72 binding domains. Protein Sci. 2006;15:1290–302. doi: 10.1110/ps.051861406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Iakhiaeva E, Wower J, Wower IK, Zwieb C. The 5e motif of eukaryotic signal recognition particle RNA contains a conserved adenosine for the binding of SRP72. RNA. 2008;14:1143–53. doi: 10.1261/rna.979508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Iakhiaeva E, Hinck CS, Hinck AP, Zwieb C. Characterization of the SRP68/72 interface of human signal recognition particle by systematic site-directed mutagenesis. Protein Sci. 2009;18:2183–95. doi: 10.1002/pro.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Iakhiaeva E, Iakhiaev A, Zwieb C. Identification of amino acid residues in protein SRP72 required for binding to a kinked 5e motif of the human signal recognition particle RNA. BMC Mol Biol. 2010;11 doi: 10.1186/1471-2199-11-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yin J, Iakhiaeva E, Menichelli E, Zwieb C. Identification of the RNA binding regions of SRP68/72 and SRP72 by systematic mutagenesis of human SRP RNA. RNA Biol. 2007;4:154–9. doi: 10.4161/rna.4.3.5428. [DOI] [PubMed] [Google Scholar]
- 154.Maity TS, Fried HM, Weeks KM. Anti-cooperative assembly of the SRP19 and SRP68/72 components of the signal recognition particle. Biochem J. 2008;415:429–37. doi: 10.1042/BJ20080569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Siegel V, Walter P. Each of the activities of Signal Recognition Particle (SRP) is contained within a distinct domain: Analysis of biochemical mutants of SRP. Cell. 1988;52:39–49. doi: 10.1016/0092-8674(88)90529-6. [DOI] [PubMed] [Google Scholar]
- 156.Ogg SC, Walter P. SRP samples nascent chains for the presence of signal sequences by interacting with ribosomes at a discrete step during translation elongation. Cell. 1995;81:1075–84. doi: 10.1016/s0092-8674(05)80012-1. [DOI] [PubMed] [Google Scholar]
- 157.Mary C, Scherrer A, Huck L, Lakkaraju AK, Thomas Y, Johnson AE, Strub K. Residues in SRP9/14 essential for elongation arrest activity of the signal recognition particle define a positively charged functional domain on one side of the protein. RNA. 2010;16:969–79. doi: 10.1261/rna.2040410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Terzi L, Pool MR, Dobberstein B, Strub K. Signal Recognition Particle Alu Domain Occupies a Defiend Site at the Ribosomal Subunite Interface upon Signal Sequence Recognition. Biochemistry. 2004;43:107–17. doi: 10.1021/bi0353777. [DOI] [PubMed] [Google Scholar]
- 159.Lakkaraju AKK, Mary C, Scherrer A, Johnson AE, Strub K. SRP keeps polypeptides translocation-competent by slowing translation to match limiting ER-targeting sites. Cell. 2008;133:440–51. doi: 10.1016/j.cell.2008.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Siegel V, Walter P. The affinity of signal recognition particle for presecretory proteins is dependent on nascent chain length. EMBO J. 1988;7:1769–75. doi: 10.1002/j.1460-2075.1988.tb03007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cline K, Henry R, Li C, Yuan J. Multiple pathways for protein transport into or across the thylakoid membrane. EMBO J. 2002;12:4105–14. doi: 10.1002/j.1460-2075.1993.tb06094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Schunemann D, Gupta S, Persello-Cartieaux F, Klimyuk VI, Jones JDG, Nussaume L, Hoffman NE. A novel signal recognition particle targets light-harvesting proteins to the thylakoid membranes. Proc Natl Acad Sci U S A. 1998;95:10312–6. doi: 10.1073/pnas.95.17.10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Franklin KE, Hoffman NE. Characterization of a chloroplast homologue of the 54-kDa subunit of the signal recognition particle. J Biol Chem. 1993;268:22175–80. [PubMed] [Google Scholar]
- 164.Li X, Henry R, Yuan J, Cline K, Hoffman N. A chloroplast homologue of the signal recognition particle subunit SRP54 is involved in the posttranslational integration of a protein into thylakoid membranes. Proc Natl Acad Sci U S A. 1995;92:3789–93. doi: 10.1073/pnas.92.9.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tu C-J, Schuenemann D, Hoffman NE. Chloroplast FtsY, Chloroplast Signal Recognition Particle, and GTP are required to reconstitute the soluble phase of light-harvesting chlorophyll protein transport into thylakoid membranes. J Biol Chem. 1999;274:27219–24. doi: 10.1074/jbc.274.38.27219. [DOI] [PubMed] [Google Scholar]
- 166.Richter CV, Trager C, Schunemann D. Evolutionary substitution of two amino acids in chloroplast SRP54 of higher plants cause its inability to bind SRP RNA. FEBS Lett. 2008;582:3223–9. doi: 10.1016/j.febslet.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 167.Funke S, Knechten T, Ollesch J, Schunemann D. A unique sequence motif in the 54-kDa subunit of the chloroplast signal recognition particle mediates binding to the 43-kDa subunit. J Biol Chem. 2005;280:8912–7. doi: 10.1074/jbc.M409992200. [DOI] [PubMed] [Google Scholar]
- 168.Hermkes R, Funke S, Richter C, Kuhlmann J, Schunemann D. The a-helix of the second chromodomain of the 43 kDa subunit of the chloroplast signal recognition particle facilitates binding to the 54 kDa subunit. FEBS Lett. 2006;580:2107–3111. doi: 10.1016/j.febslet.2006.04.055. [DOI] [PubMed] [Google Scholar]
- 169.Nilsson R, van Wikj KL. Transient interaction of cpSRP54 with elongating nascent chains of the choroplast-encoded D1 protein; ‘cpSRP54 caught in the act’. FEBS Lett. 2002;524:127–33. doi: 10.1016/s0014-5793(02)03016-8. [DOI] [PubMed] [Google Scholar]
- 170.Aldridge C, Cain P, Robinson C. Protein transport in organelles: Protein transport into and across the thylakoid membrane. FEBS J. 2009;276:1177–86. doi: 10.1111/j.1742-4658.2009.06875.x. [DOI] [PubMed] [Google Scholar]
- 171.Richter CV, Bals T, Schunemann D. Component interactions, regulation and mechanisms of chloroplast signal recognition particle-dependent protein transport. Eur J Cell Biol. 2010;89:965–73. doi: 10.1016/j.ejcb.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 172.Schunemann D. Structure and function of the chloroplast signal recognition particle. Curr Genetics. 2004;44:295–304. doi: 10.1007/s00294-003-0450-z. [DOI] [PubMed] [Google Scholar]
- 173.Jaru-Ampornpan P, Chandrasekar S, Shan S. Efficient interaction between two GTPases allows the chloroplast SRP pathway to bypass the requirement for an SRP RNA. Mol Biol Cell. 2007;18:2636–45. doi: 10.1091/mbc.E07-01-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chandrasekar S, Chartron S, Ampornpan P, Shan S. Crystal structure of the chloroplast signal recognition particle (SRP) receptor: domain arrangement modulates SRP-receptor interaction. J Mol Biol. 2008;375:425–36. doi: 10.1016/j.jmb.2007.09.061. [DOI] [PubMed] [Google Scholar]
- 175.Stengel KF, Holdermann I, Wild K, Sinning I. The structure of the chloroplast signal recognition particle (SRP) receptor reveals mechanistic details of SRP GTPase activation and a conserved membrane targeting site. FEBS Lett. 2007;581:5671–6. doi: 10.1016/j.febslet.2007.11.024. [DOI] [PubMed] [Google Scholar]
- 176.Jaru-Ampornpan P, Nguyen TX, Shan S. A distinct mechanism to achieve efficient SRP-SRP receptor interaction by the chloroplast SRP. Mol Biol Cell. 2009;20:3965–73. doi: 10.1091/mbc.E08-10-0989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Marty NJ, Rajalingam D, Kight AD, Lewis NE, Fologea D, Jumar TKS, Henry RL, Goforth RL. The membrane-binding motif of the chloroplast Signal Recognition Particle Receptor (cpFtsY) regulates GTPase activity. J Biol Chem. 2009;284:14891–903. doi: 10.1074/jbc.M900775200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Payan LA, Cline K. A stromal protein factor maintains the solubility and insertion competence of an imported thylakoid membrane protein. J Cell Biol. 1991;112:603–13. doi: 10.1083/jcb.112.4.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Reed JE, Cline K, Stephens LC, Bacot KO, Viitanen PV. Early events in the importn/assembly pathway of an integral thylakoid protein. Eur J Biochem. 1990;194:33–42. doi: 10.1111/j.1432-1033.1990.tb19423.x. [DOI] [PubMed] [Google Scholar]
- 180.Tu CJ, Peterson EC, Henry R, Hoffman NE. The L18 domain of light-harvesting chlorophyll proteins binds to chloroplast signal recognition particle 43. J Biol Chem. 2000;275:13187–90. doi: 10.1074/jbc.c000108200. [DOI] [PubMed] [Google Scholar]
- 181.Ouyang M, Li X, Ma J, Chi W, Xiao J, Zou M, Chen F, Lu C, Zhang L. LTD is a protein required for sorting light-harvesting chlorophyll-binding proteins to the chloroplast SRP. Nat Commun. 2011;2 doi: 10.1038/ncomms1278. [DOI] [PubMed] [Google Scholar]
- 182.Falk S, Sinning I. cpSRP43 is a novel chaperone specific for light-harvesting chlorophyll a,b-binding proteins. J Biol Chem. 2010;285:21655–661. doi: 10.1074/jbc.C110.132746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jaru-Ampornpan P, Shen K, Lam VQ, Ali M, Doniach S, Jia TZ, Shan S. ATP-independent reversal of a membrane protein aggregate by a chloroplast SRP subunit. Nature Struct Molec Biol. 2010;17:696–702. doi: 10.1038/nsmb.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kathir KM, Rajalingam D, Sivaraja V, Kight A, Goforth RL, Yu C, Henry R, Kumar TKS. Assembly of chloroplast signal recognition particle involves structural rearrangement in cpSRP43. J Mol Biol. 2008;381:49–60. doi: 10.1016/j.jmb.2008.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sivaraja V, Kumar TKS, Leena PST, Chang A, Vidya C, Goforth RL, Rajalingam D, Arvind K, Ye JL, Chou J, Henry R, Yu C. Three dimensional solution structures of the chromodomains of cpSRP43. J Biol Chem. 2005;280:41465–71. doi: 10.1074/jbc.M507077200. [DOI] [PubMed] [Google Scholar]
- 186.Klimyuk VI, Persello-Cartieaux F, Havaux M, Contard-David P, Schuenemann D, Meiherhoff K, Gouet P, Jones JDG, Hoffman NE, Nussaume L. A chromodomain protein encoded by the Arabidopsis CAO gene is a plant-specfic compoment of the chloroplast signal recognition particle pathway that is involved in LHCP targeting. Plant Cell. 1999;11:87–99. doi: 10.1105/tpc.11.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Delille J, Peterson EC, Johnson T, Morre M, Kight A, Henry R. A novel precursor recognition element facilitates posttranslational binding to the signal recognition particle in chloroplasts. Proc Natl Acad Sci U S A. 2000;97:1926–31. doi: 10.1073/pnas.030395197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Jonas-Straube E, Hutin C, Hoffman NE, Schunemann D. Functional analysis of the protein-interacting domains of chloroplast SRP43. J Biol Chem. 2001;276:24654–60. doi: 10.1074/jbc.M100153200. [DOI] [PubMed] [Google Scholar]
- 189.Stengel KF, Holdermann I, Cain P, Robinson C, Wild K, Sinning I. Structural basis for specific substrate recognition by the chloroplast signal recognition particle protein cpSRP43. Science. 2008;321:253–6. doi: 10.1126/science.1158640. [DOI] [PubMed] [Google Scholar]
- 190.Doyle SM, Wickner S. Hsp104 and ClpB: protein disaggregating machines. Trends in Biochemical Sciences. 2008;34:40–8. doi: 10.1016/j.tibs.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 191.Falk S, Ravaud S, Koch J, Sinning I. The C-terminus of the Alb3 membrane insertase recruits cpSRP43 to the thylakoid membrane. J Biol Chem. 2010;285:5954–62. doi: 10.1074/jbc.M109.084996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lewis NE, Marty NJ, Kathir KM, Rajalingam D, Kight AD, Daily A, Kumar TKS, Henry RL, Goforth RL. A dynamic cpSRP43-Albino3 interaction mediates translocase regulation of cpSRP targeting components. J Biol Chem. 2010;285:34220–30. doi: 10.1074/jbc.M110.160093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bals T, Dünschede B, Funke S, Schünemann D. Interplay between the cpSRP pathway components, the substrate LHCP and the translocase Alb3: an in vivo and in vitro study. FEBS Letters. 2010;584:4138–44. doi: 10.1016/j.febslet.2010.08.053. [DOI] [PubMed] [Google Scholar]
- 194.Dunschede B, Bals T, Funke S, Schunemann D. Interaction studies between the chloroplast signal recognition particle subunit cpSRP43 and the full-length translocase Alb3 reveal a membrane-embedded binding region in Alb3. J Biol Chem. 2011 doi: 10.1074/jbc.M111.250746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Jones JD, McKnight CJ, Gierasch LM. Biophysical studies of signal peptides: implications for signal sequence functions and the involvement of lipid in protein export. J Bioenerg Biomembr. 1990;22:213–32. doi: 10.1007/BF00763166. [DOI] [PubMed] [Google Scholar]
- 196.Wang Z, Jones J, Rizo J, Gierasch LM. Membrane-bound conformation of a signal peptide: A transferred nuclear overhauser effect analysis. Biochemistry. 1993;32:13991–9. doi: 10.1021/bi00213a032. [DOI] [PubMed] [Google Scholar]
- 197.Huber D, Boyd D, Xia Y, Olma MH, Gerstein M, Beckwith J. Use of thioredoxin as a reporter to identify a subset of Escherichia coli signal sequences that promote signal recognition particle-dependent translocation. J Bacteriol. 2005;187:2983–91. doi: 10.1128/JB.187.9.2983-2991.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Alamo M, Hogan DJ, Pechmann S, Albanese V, Brown PO, Frydman J. Defining the specificity of cotranslationally acting chaperones by systematic analysis of mRNAs associated with ribosome-nascent chain complexes. Plos Biology. 2011;9:e1001100. doi: 10.1371/journal.pbio.1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Peterson JH, Szabady RL, Bernstein HD. An unusual signal peptide extension inhibits the binding of bacterial presecretory proteins to the signal recognition particle, trigger factor, and the secYEG complex. J Biol Chem. 2006;281:9038–48. doi: 10.1074/jbc.M508681200. [DOI] [PubMed] [Google Scholar]
- 200.Peterson JH, Woolhead CA, Bernstein HD. Basic amino acids in a distinct subset of signal peptides promote interaction with the signal recognition particle. J Biol Chem. 2003;278:46155–62. doi: 10.1074/jbc.M309082200. [DOI] [PubMed] [Google Scholar]
- 201.Fedyukina DV, Cavagnero S. Protein folding at the exit tunnel. Ann Rev Biophys. 2011;40:337–59. doi: 10.1146/annurev-biophys-042910-155338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kramer G, Boehringer D, Ban N, Bukau B. The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat Struct Mol Biol. 2009;16:589–97. doi: 10.1038/nsmb.1614. [DOI] [PubMed] [Google Scholar]
- 203.Huber D, Rajagopalan N, Preissler S, Rocco MA, Merz F, Kramer G, Bukau B. SecA interacts with ribosomes in order to facilitate posttranslational translocation in bacteria. Mol Cell. 2011;41:343–53. doi: 10.1016/j.molcel.2010.12.028. [DOI] [PubMed] [Google Scholar]
- 204.Mariappan M, Li X, Stefanovic S, Sharma A, Mateja A, Keenan R, Hegde RS. A ribosome-associating factor chaperones tail-anchored membrane proteins. Nature. 2010;466:1120–4. doi: 10.1038/nature09296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Knoops K, Schoehn G, Schaffitzel C. Cryo-electron microscopy of ribosomal complexes in cotranslational folding, targeting, and translocation. Wiley Inbterdiscip Rev RNA. 2011 doi: 10.1002/wrna.119. [DOI] [PubMed] [Google Scholar]
- 206.Beck K, Wu L-F, Brunner J, Muller M. Discrimination between SRP- ad SecA/SecB-dependent substrates involves selective recognition of nascent chains by SRP and trigger factor. EMBO J. 2000;19:134–43. doi: 10.1093/emboj/19.1.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Buskiewicz I, Deuerling E, Gu SQ, Jockel J, Rodnina MV, Bukau B, Wintermeyer W. Trigger factor binds to ribosome-signal-recognition particle (SRP) complexes and is excluded by binding of the SRP receptor. Proc Natl Acad Sci U S A. 2004;101:7902–6. doi: 10.1073/pnas.0402231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Eisner G, Moser M, Schafer U, Beck K, Muller M. Alternative recruitment of signal recognition particle and trigger factor to the signal sequence of a growing nascent polypeptide. J Biol Chem. 2006;281:7172–9. doi: 10.1074/jbc.M511388200. [DOI] [PubMed] [Google Scholar]
- 209.Lee HC, Bernstein HD. Trigger factor retards protein export in Escherichia coli. J Biol Chem. 2002;277:43527–35. doi: 10.1074/jbc.M205950200. [DOI] [PubMed] [Google Scholar]
- 210.Ullers RS, Houben ENG, Raine A, Hagen-Jongman CM, Ehrenberg M, Brunner J, Oudega B, Harms N, Luirink J. Interplay of signal recognition particle and trigger factor at L23 near the nascent chain exit site on the Escherichia coli ribosome. J Cell Biol. 2003;161:679–84. doi: 10.1083/jcb.200302130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Zhang Y, Berndt U, Golz H, Tais A, Oellerer S, Wolfle T, Fitzke E, Rospert S. NAC functions as a modulator of SRP during the early steps of protein targeting to the ER. Mol Biol Cell. 2012 doi: 10.1091/mbc.E12-02-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Liao S, Lin J, Do H, Johnson AE. Both lumenal and cytosolic gating of the aqueous ER translocon pore are regulated from inside the ribosome during membrane protein integration. Cell. 1997;90:31–41. doi: 10.1016/s0092-8674(00)80311-6. [DOI] [PubMed] [Google Scholar]
- 213.Berndt U, Oellerer S, Zhang Y, Johnson A, Rospert S. A signal-anchor sequence stimulates signal recognition particle binding to ribosomes from inside the exit tunnel. Proc Natl Acad Sci. 2009;106:1398–403. doi: 10.1073/pnas.0808584106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Pool MR. A trans-membrane segment inside the ribosome exit tunnel triggers RAMP4 recruitment to the Sec61p translocase. J Cell Biol. 2009;185:889–902. doi: 10.1083/jcb.200807066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Dalbey RE, Wang P, Kuhn A. Assembly of bacterial inner membrane proteins. Annu Rev Biochem. 2011;80:161–87. doi: 10.1146/annurev-biochem-060409-092524. [DOI] [PubMed] [Google Scholar]
- 216.Luirink J, Samuelsson T, de Gier JW. YidC/Oxa1p/Alb3: evolutionarily conserved mediators of membrane protein assembly. FEBS Lett. 2001;501:1–5. doi: 10.1016/s0014-5793(01)02616-3. [DOI] [PubMed] [Google Scholar]
- 217.Wang P, Dalbey RE. Inserting membrane proteins: the YidC/Oxa1/Alb3 machinery in bacteria, mitochondria, and chloroplasts. Biochim Biophys Acta. 2011;1808:866–75. doi: 10.1016/j.bbamem.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 218.Samuelson JC, Chen M, Jiang F, Moller I, Wiedmann M, Kuhn A, Phillips GJ, Dalbey RE. YidC mediates membrane protein insertion in bacteria. Nature. 2000;406:637–41. doi: 10.1038/35020586. [DOI] [PubMed] [Google Scholar]
- 219.Chen M, Samuelson JC, Jiang F, Muller M, Kuhn A, Dalbey RE. Direct interaction of YidC with the Sec-independent Pf3 coat protein during its membrane protein insertion. J Biol Chem. 2002;277:7670–5. doi: 10.1074/jbc.M110644200. [DOI] [PubMed] [Google Scholar]
- 220.Chen M, Xie K, Yuan J, Yi L, Facey SJ, Pradel N, Wu LF, Kuhn A, Dalbey RE. Involvement of SecDF and YidC in the membrane insertion of M13 procoat mutants. Biochemistry. 2005;44:10741–9. doi: 10.1021/bi047418k. [DOI] [PubMed] [Google Scholar]
- 221.Facey SJ, Neugebauer SA, Krauss S, Kuhn A. The mechanosensitive channel protein MscL is targeted by the SRP to the novel YidC membrane insertion pathway of Escherichia coli. J Mol Biol. 2007;365:995–1004. doi: 10.1016/j.jmb.2006.10.083. [DOI] [PubMed] [Google Scholar]
- 222.Pop OI, Spprova Z, Koningstein G, Scheffers DJ, van Ulsen P, Wickstrom D, de Gier JW, Luirink J. YidC is required for the assembly of the MscL homopentameric pore. FEBS Lett. 2009;276:4891–9. doi: 10.1111/j.1742-4658.2009.07188.x. [DOI] [PubMed] [Google Scholar]
- 223.van der Laan M, Bechtluft P, Kol S, Nouwen N, Driessen AJ. F1F0 ATP Synthase subunit c is a substrate of the novel YidC pathway for membrane protein biogenesis. J Cell Biol. 2004;165:213–22. doi: 10.1083/jcb.200402100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Yi L, Celebi N, Chen M, Dalbey RE. Sec/SRP requirements and energetics of membrane insertion of subunits a, b, and c of the Escherichia coli F1F0 ATP synthase. J Biol Chem. 2004;279:39260–7. doi: 10.1074/jbc.M405490200. [DOI] [PubMed] [Google Scholar]
- 225.de Gier JW, Luirink J. The ribosome and YidC. New insights into the biogenesis of Escherichia coli inner membrane proteins. EMBO Rep. 2003;4:939–43. doi: 10.1038/sj.embor.embor921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kol S, Nouwen N, Driessen AJ. Mechanisms of YidC-mediated insertion and assembly of multimeric membrane protein complexes. J Biol Chem. 2008;283:31269–73. doi: 10.1074/jbc.R800029200. [DOI] [PubMed] [Google Scholar]
- 227.Xie K, Dalbey RE. Inserting proteins into the bacterial cytoplasmic membrane using the Sec and YidC translocases. Nat Rev Microbiol. 2008;6:234–44. doi: 10.1038/nrmicro3595. [DOI] [PubMed] [Google Scholar]
- 228.Oliver DC, Paetzel M. Crystal structure of the major periplasmic domain of the bacterial membrane protein assembly facilitator YidC. J Biol Chem. 2008;283:5208–16. doi: 10.1074/jbc.M708936200. [DOI] [PubMed] [Google Scholar]
- 229.Ravaud S, Stjepanovic G, Wild K, Sinning I. The crystal structure of the periplasmic domain of the Escherichia coli membrane protein insertase YidC. J Biol Chem. 2008;283:9350–8. doi: 10.1074/jbc.M710493200. [DOI] [PubMed] [Google Scholar]
- 230.Nevo-Dinur K, Nussbaum-Shochat A, Ben-Yehuba S, Amster-Choder O. Translation-independent localization of mRNA in E. coli. Science. 2011;331:1081–4. doi: 10.1126/science.1195691. [DOI] [PubMed] [Google Scholar]
- 231.Prilusky J, Bibi E. Studying membrane proteins through the eyes of the genetic code revealed a strong uracil bias in their coding mRNAs. Proc Natl Acad Sci U S A. 2009;106:6662–6. doi: 10.1073/pnas.0902029106. [DOI] [PMC free article] [PubMed] [Google Scholar]