Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum (original) (raw)
Introduction
In the molecular definition of mammalian apoptosis, experimental oncology highlighted more than two decades ago a key player, the 26 kD protein encoded by the protooncogene bcl-2. The bcl-2 gene was identified at the t(14;18) translocation of follicular B-cell lymphomas.1 At the breakpoint, a transcriptional enhancer of an immunoglobulin gene is fused to the bcl-2 gene. This enhancer/bcl-2 fusion causes large amounts of Bcl-2 to be expressed in B lymphocytes. These large amounts of Bcl-2 essentially block apoptosis in these mutant B lymphocytes and allows some B cells to survive and accumulate beyond their normal life span accumulating cell proliferation-promoting mutations.
Sequence analysis then demonstrated that Bcl-2 is the homologue of the antiapoptotic protein (CED-9) of the basic machinery of Caenorhabditis elegans, and in both normal and neoplastic tissues, its expression proved to protect cells from death by preventing or delaying apoptosis. This role is not restricted to hematopoietic (both lymphoid and myeloid) cells,2 but proved to be a general concept, holding true, for example also in the survival of central and peripheral neurons.3
These data, and the emerging role of Bcl-2 in the control of apoptosis, prompted an intense research on the site and mechanism of its action. As to the first, Korsmeyer and co-workers demonstrated in a seminal study the association of Bcl-2 with cellular membranes and in particular with mitochondrial membranes.4 This observation was pursued by a number of further studies, and the current consensus is that Bcl-2 shows a heterogeneous distribution to various cells compartments: nuclear envelope, endoplasmic reticulum (ER), outer mitochondrial membrane and, to a lesser degree, inner membrane, with also some retention within the cytosol. Interestingly, mitochondrial staining was patchy, reminiscent of mitochondrial contact zones.5, 6, 7 This complex distribution (and the putative engagement of Bcl-2 in protein complexes) suggests a pleiotropic effect, with specific and likely different functions in different membrane environments.
Major attention was obviously drawn to mitochondria, after increased permeability of the outer mitochondrial membrane emerged as an early step of intracellular death signalling. This mechanism allows apoptogenic factors such as cytochrome c to be released into the cytosol where they contribute to caspase activation.8 As antiapoptotic members of the Bcl-2 family prevent the increase in membrane permeability and protect cells from various death insults, it has been assumed that Bcl-2 family members primarily regulate mitochondrial integrity, and thus the crucial commitment step in many apoptotic routes.9
However, in the recent years also the other potential sites of Bcl-2 action regained interest, for at least two reasons. The first is that mitochondrial apoptotic involvement could depend on signals that originate from other cell compartments. Namely, Ca2+ rises triggered by release from the ER could induce and/or play a facilitatory role in the apoptotic changes of mitochondria,10 as discussed later in greater detail. Second, apoptotic signalling can bypass mitochondria and even occur independently of caspase activation, but still be controlled by Bcl-2. Specifically, the alteration of ER functions could per se protect cells from apoptosis. The complexity of the scenario is supported by apparently conflicting results. Serum starvation induced apoptosis in MDCK cells expressing Bcl-2 targeted to the ER but not to the mitochondria, while on the contrary the protein located to the ER was more efficient in protecting from apoptosis Rat-1/myc cells.11 Lee et al.12 reported that in Rat-1 fibroblasts, Bcl-2 targeted to the ER was able to inhibit Myc- but not etoposide-induced apoptosis. McCullough et al.13 reported that overexpression of gadd153/chop sensitizes cells to ER stress, with ensuing downregulation of Bcl-2 expression, depletion of cellular glutathione, and enhanced production of reactive oxygen species, and restoration of Bcl-2 fully reverts the phenotype.
Thus, Bcl-2 appears capable of managing life-or-death decisions at several intracellular sites, via more than one mechanism and perhaps even in a cell-type-specific manner. In this review, we will analyse the functions of Bcl-2 outside of the mitochondria (extensively described in other reviews presented in this special issue) focusing our attention on the ER and calcium homeostasis.
Bcl-2 and Ca2+: Establishing the Link
Cell death has always been known to be one of the numerous cellular events triggered by increases in intracellular Ca2+ concentration evoked by physiological or pathological stimuli. Classically, this has been associated to necrosis, that is the catastrophic derangement of cell integrity and function following different types of cell injury and leading to activation of Ca2+-activated hydrolysing enzymes. A typical example is excitotoxicity, in which glutamate-dependent hyperstimulation leads neurons to death.14, 15 The link to apoptosis was appreciated more recently, and the analysis of Bcl-2 has been a crucial element in attributing a role for Ca2+ in apoptosis. When studying the complex mechanism of action of this oncogene, the possibility that it could act, at least in part, by modulating ion fluxes was suggested by its intracellular distribution, that is to organelles playing a key role in Ca2+ homeostasis, the ER and mitochondria.16 Two important contributions corroborated this hypothesis, prompting further research on the topic. First, Bcl-2 and related proteins were shown to act as cation channels. Specifically, the three-dimensional structure of the Bcl-2 homologue, Bcl-XL, bears a strong resemblance to some pore-forming bacterial toxins.17 Indeed, Bcl-2 itself was shown to form cation channels of low selectivity in artificial lipid bilayers,18 although later studies showed that the channel mostly conducts sodium and not calcium.19
Second, Bcl-2 expression was shown to modify the changes in Ca2+ homeostasis evoked by IL-3 deprivation in haematopoietic cell lines.20 Specifically, Bcl-2 expression was shown to prevent the reduction in cytoplasmic Ca2+ levels and in the size of the TG-releasable pool that was observed upon withdrawal of IL-3. In lymphoma cells, Bcl-2 overexpression was reported to decrease the size of the TG-releasable pool, but not to prevent capacitative Ca2+ entry of extracellular calcium. Further work by the same authors showed that Bcl-2 prevents ER depletion at low extracellular Ca2+ and maintains Ca2+ uptake in TG-treated cells (reviewed in Distelhorst and Shore21). Overall, the data, although complex and hard to reconcile into a unifying mechanism, unquestionably associated Bcl-2 and ER Ca2+ homeostasis, and prompted a number of researchers to investigate directly, as soon as suitable probes became available, the effect of Bcl-2 on Ca2+ levels in the ER lumen and the relevance of this effect for the antiapoptotic activity of the protein.
To further increase the interest in Bcl-2 effects on ER Ca2+ homeostasis, in the same years the role of mitochondria in calcium signalling was greatly re-evaluated.22 Also in this case, the development of novel, protein-based probes allowing the specific measurement of Ca2+ concentration within the mitochondrial matrix (mitochondrial matrix Ca2+ concentration ([Ca2+]m)) was decisive. With new luminescent23 or green fluorescent protein (GFP)-based fluorescent24 probes, in virtually all cell types a large [Ca2+]m increase was shown to parallel the cytosolic increases occurring upon cell stimulation. In this process, the ER (and its interactions with mitochondria) were shown to play a key role. Indeed, at least in cells in which rapid Ca2+ responses depend on the opening of ER Ca2+ channels, the strategic location of mitochondria close to the source of the cytosolic Ca2+ concentration ([Ca2+]c) rise (the ER) allows them to be exposed to microdomains of high [Ca2+], that meet the low affinity of their transporters and allows the rapid and large accumulation of the cation in the matrix.25, 26
Mitochondrial Ca2+ accumulation, in turn, can induce radically different effects, and participate in functionally decoding the Ca2+-mediated signals. On the one hand, by stimulating Ca2+-dependent enzymes27, 28, 29 or metabolite carriers30 it can stimulate aerobic metabolism31 and enhance ATP production.32 On the other hand, Ca2+ may synergize with apoptotic mediators and induce large-scale morphological changes of the organelle, possibly by favouring the opening of the large-conductance channel known as permeability transition pore (PTP),33 and thus induce the release of mitochondrial proapoptotic factors. Not surprisingly, loading of Ca2+ in the matrix, and the opening of PTP, has been implicated in the pathogenesis of numerous human disorders, stemming from heart damage upon reperfusion to the pathogenesis of muscular dystrophies.34, 35
Direct Measurements of Endoplasmic Reticulum Ca2+ Concentration ([Ca2+]er) in Cells Overxpressing Bcl-2
The development of molecular biology techniques (which enable the modification and expression of exogenous cDNA in heterologous cell types), has been responsible in recent years for the widespread use of protein probes by cell biologists. As far as Ca2+ is concerned, the first probe of this type was aequorin, but then the molecular engineering of GFP yielded gene-encoded probes endowed with strong fluorescence and suitable for single-cell imaging studies. In both cases, low-affinity, ER-targeted probes were developed that allowed the direct monitoring of ER Ca2+ levels (for methodological aspects, and the advantages and pitfalls of each class of recombinant probes, we refer to a recent review by Rudolf et al.36).
Using these different probes, three groups have addressed the issue of Bcl-2 effects on ER Ca2+ levels, our own and those of KH Krause and RY Tsien.37, 38, 39 We utilized an ER-targeted aequorin chimera40 and transiently coexpressed it with Bcl-2 in HeLa cells. No toxicity due to Bcl-2 transient overexpression was observed, in distinction to previous reports on the use of a Bcl-2-GFP chimera.41 Rather, the cells overexpressing Bcl-2 displayed an enhanced survival upon ceramide treatment,42 in agreement with previous reports.43, 44 Bcl-2 showed the expected heterogeneous distribution, most of it being located on the ER membrane. As to Ca2+ homeostasis (Figure 1), we detected a ∼30% reduction in the Ca2+ levels within the agonist-sensitive Ca2+ stores (i.e. both the ER and the Golgi apparatus). Consequently, stimulus-dependent [Ca2+] increases were reduced both in the cytoplasm and in the mitochondria.37 The studies also gave some insight into the mechanism of this alteration: no difference in the initial rate of uptake was detected, thus suggesting an increased leak from the organelles, rather than a reduction in the activity of the ER Ca2+ ATPase (SERCAs). Direct measurement of the leak rate supported this view.
Figure 1
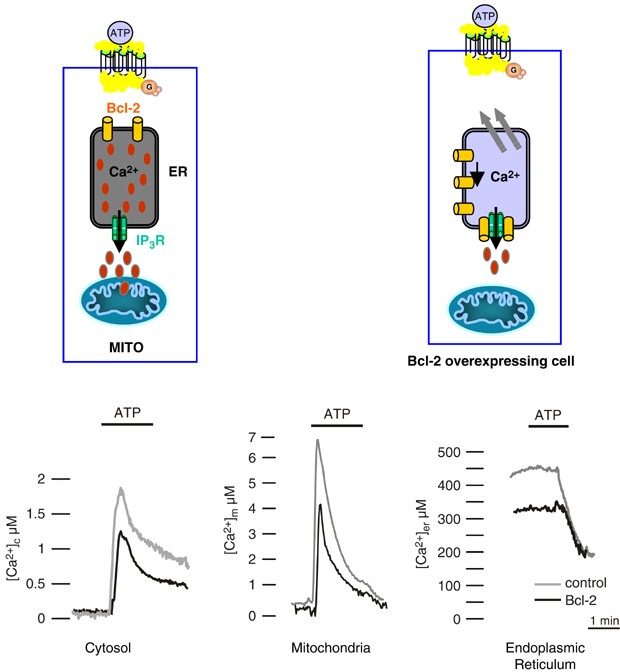
Subcellular analysis of Ca2+ homeostasis in control and Bcl-2-overexpressing HeLa cells. The traces show representative [Ca2+] measurements performed in the cell cytoplasm, mitochondria and endoplasmic reticulum. Grey: control cells; black: Bcl-2-overexpressing cells. To monitor [Ca2+] in the different compartments of transiently transfected Bcl-2 or control cells, HeLa cells were cotransfected with specifically targeted aequorin chimeras.40 Cells were perfused with Krebs–Ringer saline solution and challenged, where indicated, with 100 _μ_M ATP
The other two groups employed GFP-based probes. Krause and co-workers utilized an ER-targeted ‘cameleon’ and reported a lower steady-state [Ca2+]er in Bcl-2-overexpressing HeLa cells and an increased leak rate upon thapsigargin blockage of SERCA pumps.38 More recently, Tsien and co-workers developed an optimized cameleon probe, in which the Ca2+-binding site of calmodulin and the calmodulin-binding peptide were coordinately mutated, and the affinity was suitable to the concentration range of the ER lumen. With this probe, that does not perturb calmodulin-dependent signalling (in distinction to the classical probes based on the recombinant expression of wild-type calmodulin), the notion of a reduced Ca2+ level in the ER due to increased Ca2+ leak was confirmed. Interestingly, these authors also showed that the green tea compound epigallocatechin gallate, known to bind to Bcl-2 and induce apoptosis in Bcl-2-expressing cells, inhibits Ca2+ leakage and restores [Ca2+]er values.39
Overall, the three studies provide an amazingly coherent picture: despite the use of radically different probes, with different detection and calibration procedures (which may account for the significant discrepancy in the absolute [Ca2+] values), the direct measurement of [Ca2+]er reveals in all cases a 20–40% decrease of the resting values. Conceptually, similar data were also obtained with other unrelated antiapoptotic proteins. The most striking example was provided by an oncogene recently described in a human hepatocarcinoma. This oncogene is generated by the integration of the hepatitis B (HBx) virus genome in the gene encoding the protein SERCA1. Viral activation was shown to _cis_-activate SERCA1 chimeric transcripts with splicing of exon 4 and/or exon 11. Splicing of exon 11 creates a frameshift and a premature stop codon in exon 12. The encoded protein lacks most of the cytosolic N and P domains and critical Ca2+-binding regions of the transmembrane region. This protein is incapable of active Ca2+ pumping,45 and is causally involved in the neoplastic phenotype. Although the molecular mechanism of this oncogene has not been explained yet, it may be speculated that the mutated SERCA could either interfere with the activity of endogenous pumps and/or it could act as a Ca2+ leak pathway from the ER. These data are consistent with the recent observations that overexpression of SERCA in HeLa cells increases the susceptibility of cells to apoptotic agents.42, 46
The final, unquestionable proof of the relationship between Bcl-2 and Ca2+ homestasis came again from the elegant work of Stan and his group. In a seminal paper published in Science, Korsmeyer and co-workers demonstrated that cells from mice in which the genes for Bax and Bak (proapoptotic proteins that associate with mitochondria and initiate organelle dysfunction, but are also localized to the ER) are endowed with partially depleted ER stores, suggesting that these proteins directly counteract the effect of Bcl-2 on calcium signalling (and thus the latter protein, unopposed in the Bax/Bak knockouts (KO), allows lower [Ca2+]er levels to be attained). In support of this possibility, siRNA silencing also of Bcl-2 restored [Ca2+]er values similar to those of controls.47 Along these lines, we then showed that early after overexpression of Bax in HeLa cells [Ca2+]er levels are higher, whereas at later stages (and progression into the apoptotic phenotype) the difference with control cells becomes virtually undetectable.48 The common scientific interest was also the opportunity for us to meet Stan Korsmeyer and discuss science with him. The friendly and stimulating discussions, as well as ideas and speculations on ‘the bigger picture’, turned out even more exciting that our initial enthusiasm that our data and initial hypothesis was supported by the main authority in the field (and his powerful transgenic models). We miss these discussions, as well as the collaborative work that we had planned but did not have time to carry out.
In this context, the observation that Bcl-2 also downregulates Ca2+ influx through the plasma membrane is not surprising. In principle, depletion of the ER Ca2+ store could bring to activation of capacitative Ca2+ influx,49 and thus induce a prolonged [Ca2+]c elevation (i.e. a potentiation of Ca2+-mediated effects on apoptosis). Indeed, ER depletion of comparable degree was reported to cause a substantial activation (over 50%) of store-dependent Ca2+ influx.50 Conversely, Bcl-2 expression markedly reduces the [Ca2+]c increase induced by Ca2+ readdition to cells in which the Ca2+ store had been fully depleted by SERCA blockers.37 This effect, that may represent a long-term adaptation to lower [Ca2+]er levels, is most likely due to reduction of the number of functional channels in the plasma membrane.51 This route appears to be followed also by other antiapoptotic proteins. In analysing the mechanism of neuroendocrine differentiation (that incidentally is a common hallmark of a variety of carcinomas), Vanoverberghe et al.52 showed that in LNCaP cells the same Ca2+ signalling alteration (partial ER depletion and reduction of the capacitative Ca2+ current) was observed upon Bcl-2 expression and upon induction of neuroendocrine differentiation, although in the latter case different molecular mechanisms may be operative.
The general picture, however, is far from being fully unraveled. Indeed, in the same years a number of reports appeared which did not support the notion that Bcl-2 reduces ER Ca2+ levels. The largest body of work was carried out by the group of Distelhorst19, 41, 53, 54, 55 that in recent papers reported that the size of ionomycin-releasable intracellular Ca2+ pools was the same in controls and Bcl-2-expressing cells.56 Work by other groups support this view. In rat kidney cells57 and in a murine hypothalamic cell line,58 after treatment with thapsigargin no difference between control and Bcl-2-expressing cells in the increase of [Ca2+]c were observed. The obvious implication is that the size of the ER Ca2+ pool is not affected. Further complexity is added by the results of Kuo and co-workers who observed a larger [Ca2+]c increase upon thapsigargin treatment in Bcl-2 overexpressing compared to control cells. The authors suggest that the increase in the amount of Ca2+ released in Bcl-2-overexpressing cells is the consequence of the up regulation of SERCA gene expression and a direct activation possibly due to protein–protein interaction of Bcl-2 with SERCA.59 Finding a reason for these discrepancies is not easy. One could argue that most of these studies monitor [Ca2+]er indirectly (i.e. through the cytosolic peak triggered by physiological or pharmacological emptying of the ER). However, these approaches were employed also in the studies by the other groups, and a difference in the size of the pool was detected.60 A unifying explanation therefore needs to imply that the situation differs in various cell systems, and thus cell-type-specific regulatory mechanisms may control the amplitude, if not the occurrence, of the Bcl-2 effect. The analysis of the molecular mechanism of the Bcl-2 effect may support such a possibility. Indeed, as we will discuss in detail later, recent data strongly argue for the possibility that isoform-specific interaction with the inositol trisphosphate (IP3) receptor is at the base of the Bcl-2 effect. In this case, it is reasonable to assume that the expression profile of inositol trisphosphate receptors (IP3Rs) or modulatory proteins can tune the responsiveness of cells to the signalling effects of Bcl-2.
The Correlation of the Calcium-Signalling Alteration with Sensitivity to Apoptosis
Although the demonstration of partial ER emptying upon Bcl-2 expression and of the opposite effect of proapoptotic Bcl-2 family members was strongly arguing for a functional role of the signalling alteration, the concept needed experimental verification, that was obtained by different groups. In our own work, we showed that if the Bcl-2 effect on [Ca2+]er was replicated by different pharmacological and molecular approaches (in the absence of the oncoprotein) the cells were protected from ceramide, a Bcl-2-sensitive apoptotic stimulus, while treatments that increased [Ca2+]er had the opposite effect.42 A similar picture emerged from the study with the Bax/Bak KO. In those cells, when the ER Ca2+ levels were restored by recombinantly overexpressing the SERCA, not only mitochondrial Ca2+ uptake in response to stimulation was re-established, but the cells regained sensitivity to apoptotic stimuli such as arachidonic acid, C2-ceramide and oxidative stress.47 These results are in keeping with previous work by Ma et al.46, demonstrating that SERCA overexpression in Cos cells causes ER Ca2+ overload and increases spontaneous apoptosis. Higher [Ca2+]er levels imply a larger amount of Ca2+ that can be released into the cytosol (and taken up by mitochondria), but also a different regulation of Ca2+-sensitive luminal processes of the ER (such as protein post-translational modifications and sorting). In order to verify which is the real target of the signalling modulation, we and others have acted on the luminal buffer, the low-affinity Ca2+-binding protein calreticulin and demonstrated that the protective effect depends on the decrease of the releasable Ca2+ pool. In calreticulin overexpressing cells, in which the amplitude and duration of Ca2+ signals from ER lumen towards cytosol are enhanced61 without changing [Ca2+]er,62 cell viability is drastically reduced upon ceramide treatment.63 Conversely, cell lines derived from calreticulin KO, that show a marked decrease in ER Ca2+ release upon cell stimulation, are more resistant to apoptosis.64
The importance of the size of the releasable pool, that is the concept that the key determinant is the net amplitude of the cytosolic rise (as well as of the mitochondrial loading, as we will discuss later) was further reinforced by the study of two viral proteins. The antiapoptotic Coxsackie viral protein 2B was shown to reduce ER Ca2+ levels.65 On the contrary the proapoptotic protein of HBx augments the cytosolic Ca2+ signals evoked by InsP3-linked agonists. However, this is not due to ER Ca2+ overload, as the steady-state [Ca2+]er levels are identical to those of mock-transfected cells.66 The mechanism leading to cell death is similar to what previously reported in staurosporin-treated neurons: the Ca2+ extrusion capacity of the cell is impaired by caspase-3-dependent cleavage of the plasma membrane Ca2+ ATPase (PMCA).67 Indeed in HBx-transfected cells there is caspase-3 activation and PMCA cleavage, and recombinant expression of a noncleavable PMCA mutant both restored the Ca2+-signalling patterns and amplitude to those of control cells and reduced the apoptotic efficiency of the viral protein. PMCA represents a very effective target for enhancing Ca2+ responses, because it represents the most powerful route allowing the rapid return of [Ca2+]c to basal level.66 Along the same lines, the Na+/Ca2+ transporter type 1 is also cleaved by caspase-3 in cerebellar granule neurons undergoing apoptosis.68
Two final aspects require attention. Firstly, the idea that a [Ca2+]er decrease protects cells from the effect of apoptotic agents may appear in contrast with the established notion that treatment of cells with SERCA inhibitors (thapsigargin, tBuBHQ, and cyclopiazonic acid) is followed by apoptosis.69 It should be remembered, however, that the effect on [Ca2+]er is radically different in the two conditions. Upon SERCA blockage the Ca2+ depletion is complete and rapid, while in Bcl-2-transfected cells the drop in [Ca2+]er is modest and develops slowly. A drastic reduction in the level of [Ca2+]er might interfere with the basic ER functions (e.g. the regulation of ER protein folding and chaperone interactions), and thus cause a stress response that leads to cell death. The second aspect that needs to be remembered is that calcium is not only associated to cell death, but mediates cell-specific activities and responsiveness to mitogenenic stimuli. Thus, a modification of Ca2+-signalling patterns may have a pleiotropic effect, if both types of signalling are affected. Such a possibility was directly investigated by the Distelhorst group, that analysed in the very same system (T cells) the functional consequences of Bcl-2-dependent reduction of Ca2+-mediated signals. Interestingly, they could show that Ca2+ signals elicited by maximal T-cell receptor stimulation (such as those triggered by high concentrations of anti-CD3 antibody) were significantly reduced in Bcl-2-expressing cells, whereas prosurvival Ca2+ signals induced by weak anti-CD3 stimulation were unaffected, possibly due to different requirement (or regulation) of the IP3R.56
Identifying the Effector: a Primary Role for Mitochondria?
The data so far summarized indicate that, through a reduction in ER Ca2+ levels, upon expression of Bcl-2 (and other antiapoptotic proteins) cellular Ca2+ signals elicited by physiological and pathological stimuli are reduced, and this exerts a protective effect in some apoptotic routes. The search for the effector systems is complex, and will most likely identify molecular mechanisms operating in different cell compartments. Namely, there is no doubt that a reduction in cytosolic Ca2+ signals will affect the activity of enzymes that have been shown to modulate the apoptotic process. To name a few, Ca2+-dependent proteases, such as calpains, have been shown to be active in cell death pathways, and the ablation of their genes associated to muscle-degenerative disorders. As to kinases and phosphatases, a broad literature associates calcineurin and the different isoforms of PKC to the modulation of apoptosis (with specific, even opposite effect of different isoforms).70, 71
In the past years, however, major attention has been devoted to mitochondria, triggered by two important discoveries. The first was the demonstration of the release during apoptosis of a specific subset of mitochondrial proteins into the cytosol. These proteins, that include cytochrome c, Smac/DIABLO and AIF, form with effector caspases a macromolecular machinery (the apoptosome) that precipitates cells into apoptotic death.8 The second observation was the functional analysis of a large-conductance mitochondrial channel, the PTP, that is activated by a variety of toxic challenges and cell signals (including increases in [Ca2+]m) and causes mitochondrial swelling and fragmentation. Although the molecular definition of these process is still uncertain, these morphological alterations may represent an efficient route for the release of proapoptotic proteins. Indeed, PTP opening, implicated in many types of cellular dysfunction and death, was a likely candidate for Ca2+-dependent effector of apoptosis.72, 73
We and others thus verified whether apoptotic challenges lead to mitochondrial alterations, and whether Ca2+ was involved in the process. In HeLa cells we observed Ca2+ release from the ER upon ceramide treatment and loading into mitochondria (Figure 2). As a consequence, organelle swelling and fragmentation were detected, that were parallel by release of cytochome c. These changes were prevented by Bcl-2 expression as well as experimental conditions that preserved [Ca2+]er.42 PTP opening in ceramide-dependent apoptosis was directly demonstrated by Hajnoczky and co-workers who could demonstrate that the lipid mediator facilitates PTP opening. In this case, ceramide acts as a ‘mitochondrial sensitizer’, that thus transforms physiological IP3-mediated signals into inducers of apoptosis33(Figure 2). A similar picture emerged with other apoptotic inducers. HBx caused, both in HeLa and hepatic cell lines, a dramatic fragmentation and swelling of the mitochondrial network, that was inhibited (together with the apoptotic efficacy) by the PTP blocker cyclosporin A.66 As to Bax, its primary effect on Ca2+ homeostasis was, early upon expression, an increased ER Ca2+ loading, with consequent potentiation of mitochondrial Ca2+ responses, then the cells progressed into an apoptotic phenotype (that included drastic reductions of the cytosolic and mitochondrial Ca2+ responses upon challenging of cells with stimuli causing ER Ca2+ release). In this later phase, no difference became detectable between the steady-state [Ca2+]er levels of controls and Bax-expressing cells. Under those conditions, two features dominated the general picture of Ca2+ signalling. The first was a drastic perturbation of mitochondrial three-dimensional structure and of key functional parameters, including mitochondrial membrane potential and the Ca2+ uptake capacity of the organelle.48 This effect also provides a possible explanation for second feature, the gradual decrease in ER Ca2+ loading: reduced ATP provision to the SERCA pumps could gradually impair Ca2+ (re)accumulation into the ER, thus bringing the [Ca2+]er steady-state levels to those of a control cell. An alternative possibility stems from the recent demonstration that the IP3R includes a consensus site for caspase-3 cleavage and the cleaved form of the receptor exhibits increased leakiness and reduced sensitivity to IP3.74, 75 In turn, a smaller Ca2+ release results in the reduction of the amplitude of cytosolic Ca2+ responses (an effect that cooperates with the direct effect on mitochondria in causing the dramatic, 30–50% reduction of mitochondrial [Ca2+] peaks).
Figure 2
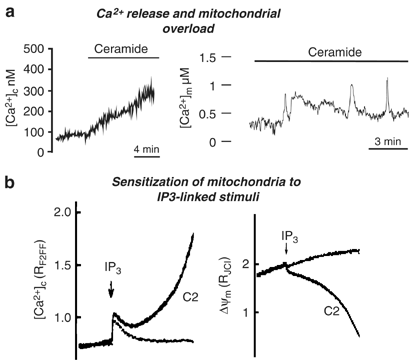
Possible mechanisms of ceramide action on Ca2+ homeostasis and mitochondrial function. (a) Ceramide can induce leak of stored Ca2+ from the ER. Depletion of the ER Ca2+ content activates capacitative calcium influx with further [Ca2+]c increase and mitochondrial Ca2+ loading as measured with aequorin-based probes (reproduced with permission from Pinton et al.63). In turn this causes major morphological alterations of mitochondria, and thus release of proapoptotic factors to the cytoplasm; (b) ceramide can induce sensitization of mitochondria to IP3-linked stimuli (reproduced with permission from Szalai et al.33). Simultaneous measurements of mitochondrial membrane potential (ΔΨm) measured with TMRM and [Ca2+]c measured using fura2F/FFA during IP3R stimulation in control and ceramide (C2)-treated cells are shown
In summary, Bcl-2 and other antiapoptotic proteins reduce ER Ca2+ levels, and consequently they moderate the efficacy of apoptotic mediators that use Ca2+ signals (and the involvement of mitochondria as downstream effectors) as a potentiation/commitment factor. Conversely, Bax enhances the loading of the ER Ca2+ store, and thus boosts the Ca2+ load to which the apoptotic effector systems (including mitochondria) are exposed upon physiological and/or pathological challenges. This effect of Bax coincides with gross perturbation of mitochondrial structure and function, and finally, later in apoptotic progression, to the development of an altered signalling phenotype, that includes impaired ER Ca2+ release upon cell stimulation, and thus reduction of cellular Ca2+ signals. What is unclear from these studies is whether the ability of Bcl-2 and Bax to regulate ER [Ca2+] is an intrinsic property of these proteins or rather the outcome of a regulatory effect on other proteins.
The Molecular Mechanism
Bcl-2 channel activity
The molecular mechanism by which Bcl-2 alters ER Ca2+ leakiness channels is still unresolved. In light of the evidence that Bcl-2 may be an ion channel,17, 18 the simplest explanation for the increased passive leakage observed in Bcl-2-overepressing cells would be a higher density of Bcl-2 channels. In other words, Bcl-2 could facilitate Ca2+ leakage from the ER by forming a channel, either by oligomerizing or by interacting with other members of the Bcl-2 family. The conceptual problem with this explanation (strongly supported by the demonstration of ion conductance in lipid bilayers) is that also Bax has structural similarities with pore-forming bacterial toxin and thus can in principle form ion channels in artificial membranes. However, Bax augments, not reduces [Ca2+]er. One could argue that the channel properties of the two pore-forming domains may be different (e.g. for the presence of acidic residues in Bcl-2 where basic ones are present in Bax), thus accounting for different ion selectivity of the channel activity of the two proteins (reviewed in Schendel et al.17). Specifically, the predicted fifth and sixth _α_-helices of Bcl-2 and Bax are hypothesized to directly participate in channel formation. These _α_-helices are positioned in the core of these proteins and are believed to be inserted into the lipid bilayer with the loop connecting _α_5 and _α_6 presumably protruding on the other side of the membrane. Indeed, deletion of the _α_5-_α_6 regions from Bcl-2 abolishes its ability to form ion channels in synthetic membranes in vitro.18, 76 To verify the role of the presumed channel-forming _α_5–6 helices of Bcl-2 and Bax in Ca2+ mobilization were used two chimerical constructs, in which the putative pore domains of the proteins were mutually swapped.77 The results (Figure 3) showed that when the putative pore-forming domain of Bax replaced that of Bcl-2 in the Bcl-2 polypeptide the chimeric protein was still capable of reducing [Ca2+]er. Conversely, the Bax chimera including the putative pore-forming domain of Bcl-2 did not significantly alter [Ca2+]er, similarly to the wild-type protein. Accordingly, the two chimeras retained the effects on apoptosis of the original protein: the Bax protein with Bcl-2 (_α_5–6) helices is proapoptotic, and causes perturbation of mitochondrial structure and function (including the drastic reduction of [Ca2+]m responses), whereas the Bcl-2 chimera with Bax (_α_5–6) protects against stimuli, such as ceramide, acting through a calcium- and mitochondria-dependent pathway.48 The general picture emerging from these results is that Bax and Bcl-2 both have an effect on Ca2+ signalling, that does not depend on the region proposed to form an ion channel.
Figure 3
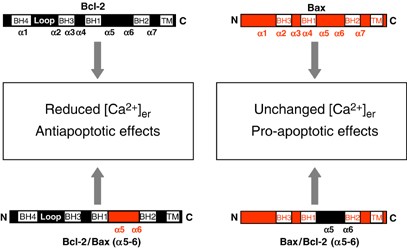
Schematic representation of the Bcl-2 and Bax chimera and of the putative regions of the encoded polypeptide. The predicted positions of the _α_-helical regions within the human Bcl-2 and Bax proteins are shown. Bcl-2/Bax(_α_5–6) corresponds to a Bcl-2 construct in which the putative pore-forming domain was replaced with that of Bax. Bax/Bcl-2(_α_5–6) corresponds to a Bax construct in which the putative pore-forming domain was replaced with that of Bcl-2. BH, Bcl-2 homology domains; TM, transmembrane domaine
Direct molecular interaction with ER channels
An alternative, intriguing possibility that links Bcl-2 to cellular Ca2+ signalling through a mechanism independent of the pore-forming domain, is that Bcl-2 could interact with and alter the function of an endogenous release channel or pore-forming protein in the ER (Figure 4). Recently, strong experimental evidence was obtained indicating that Bcl-2 and related antiapoptotic proteins regulate the most logical target, that is the IP3R, although two conceptually different mechanisms are proposed. The first article was contributed by Stan Korsmeyer's group. Stan and co-workers demonstrated that Bcl-2 controls the phosphorylation state of type I IP3R, that in turn regulates the leak rate through the channel. In support of this view, the [Ca2+]er reduction of Bax/Bak KO was reversed by siRNA silencing of IP3R-I.78 More recently, the groups of Kevin Foskett and Craig Thompson demonstrated that Bcl-XL directly binds to the IP3R and in single-channel electrophysiology experiments with isolated nuclei both the addition and the recombinant expression of Bcl-XL sensitized the IP3R to low agonist doses. Recombinant expression (or addition to the patch pipette) of Bax prevented the effect of Bcl-XL, both in terms of its binding to the IP3R and of capacity of modifying the sensitivity to IP3.79 The results differ from a previous paper by the Distelhorst group that showed that Bcl-2 reduces the opening probability of IP3Rs inserted into lipid bilayers.53 As to the mechanism, the Foskett–Thompson paper proposes a direct modulatory role on IP3R (through protein–protein interaction between the two proteins) rather than a phosphorylation event, based on the timing after Bcl-XL addition and the experimental conditions employed (nonpermissive for phosphorylation). Work needs to be carried out to verify whether the different results of these papers reflect true experimental discrepancies or functional complexity. Indeed, not only the Bcl-2 family members could differ in the signalling mechanism, but the variability between cell lines in IP3R levels, isoform expression (given that IP3RI is proposed to by phosphorylated) and basal levels of IP3 (key regulatory determinant in the Foskett–Thompson paper) could account for the apparently conflicting results. Such a complexity could also be reflected in the previously discussed discrepancies in the Ca2+ measurements. Where all papers agree in on the important concept that the IP3R is at the centre of the stage, and Bcl-2 hits the main route for Ca2+ release from intracellular stores. This observation well fits with the observation that Bcl-2 alters Ca2+ oscillations triggered by agonist acting by IP3R stimulation39 and with studies reporting a correlation between low expression of IP3R and inhibition of apoptosis.80 Finally, the Foskett–Thompson paper, by looking at sensitivity to apoptosis of DT40 cells in which all three IP3R isoforms had been deleted, shows that basal activation of the IP3R not only partially depletes the Ca2+ store but also provides a ‘survival’ signal per se (possibly by activating mitochondrial metabolism), given that the KO cells are more sensitive to apoptotic challenges than the wild-type cells.79 Also in this case, given the robust evidence for the role of mitochondrial Ca2+ uptake in facilitating PTP opening and proapoptotic mitochondrial changes, it seems fair to conclude that Bcl-2 globally reprogrammes ER Ca2+ release and mitochondrial Ca2+ loading, as necessary for optimally activating apoptosis (in terms of metabolic requirements as well as prompt release of caspase cofactors into the cytosol).
Figure 4
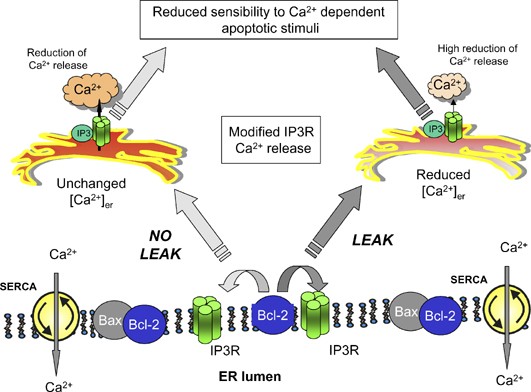
Schematic model of Bcl-2 effects on Ca2+ homeostasis. Two nonmutually exclusive mechanisms are presented. In the first (right part) Bcl-2 interacting with the IP3R induces Ca2+ leakage from the ER, in turn causing a significant reduction of [Ca2+]er and a consequent impairment of Ca2+ release from ER after stimulation. In the second scenario (left part) Bcl-2 interacting with the IP3R (possibly different subtypes) reduces the sensitivity to IP3-linked stimuli. In both cases Bcl-2 and other antiapoptotic proteins, can reduce ER Ca2+ release, and consequently moderate the efficacy of apoptotic mediators that use Ca2+ signals as a potentiation/commitment factor
Conclusions
A broad series of data now allows to state that Bcl-2 expression interferes with cellular Ca2+ signals, most likely by acting on the IP3R and thus modifying Ca2+ leakage in resting conditions (and thus the steady-state [Ca2+]er) and its release kinetics upon stimulation. In turn, this affects the efficacy of apoptotic challenges by influencing the sensitivity of Ca2+ effectors, such as mitochondria. Despite the significant advancements of the last years, much remains to be understood, including the final assessment of the molecular mechanism of the Bcl-2 effect and the significance of this process for apoptosis in vivo. However, these data already strengthen the view that also apoptotic cell death belongs to the numerous cell functions modulated by Ca2+ ions and thus add a well-known pharmaceutical target to the possibilities for modulating the route to cell degeneration.
Abbreviations
ER:
endoplasmic reticulum
[Ca2+]m:
mitochondrial matrix Ca2+ concentration
[Ca2+]er:
endoplasmic reticulum Ca2+ concentration
[Ca2+]c:
cytosolic Ca2+ concentration
GFP:
green fluorescent protein
PTP:
permeability transition pore
SERCAs:
ER Ca2+ ATPasi
PMCA:
plasma membrane Ca2+ ATPase
IP3:
inositol trisphosphate
IP3R:
inositol trisphosphate receptor
References
- Tsujimoto Y, Finger LR, Yunis J, Nowell PC and Croce CM (1984) Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226: 1097–1099.
Article CAS PubMed Google Scholar - Vaux DL, Cory S and Adams JM (1988) Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335: 440–442.
Article CAS PubMed Google Scholar - Holm K and Isacson O (1999) Factors intrinsic to the neuron can induce and maintain its ability to promote axonal outgrowth: a role for BCL2? Trends Neurosci. 22: 269–273.
Article CAS PubMed Google Scholar - Hockenbery D, Nunez G, Milliman C, Schreiber RD and Korsmeyer SJ (1990) Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348: 334–336.
Article CAS PubMed Google Scholar - Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W and Reed JC (1993) Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 53: 4701–4714.
CAS PubMed Google Scholar - Akao Y, Otsuki Y, Kataoka S, Ito Y and Tsujimoto Y (1994) Multiple subcellular localization of bcl-2: detection in nuclear outer membrane, endoplasmic reticulum membrane, and mitochondrial membranes. Cancer Res. 54: 2468–2471.
CAS PubMed Google Scholar - de Jong D, Prins F, van Krieken HH, Mason DY, van Ommen GB and Kluin PM (1992) Subcellular localization of bcl-2 protein. Curr. Top. Microbiol. Immunol. 182: 287–292.
CAS PubMed Google Scholar - Kroemer G and Reed JC (2000) Mitochondrial control of cell death. Nat. Med. 6: 513–519.
Article CAS PubMed Google Scholar - Lucken-Ardjomande S and Martinou JC (2005) Regulation of Bcl-2 proteins and of the permeability of the outer mitochondrial membrane. C. R. Biol. 328: 616–631.
Article CAS PubMed Google Scholar - Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T and Rizzuto R (2002) A role for calcium in Bcl-2 action? Biochimie 84: 195–201.
Article CAS PubMed Google Scholar - Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B and Andrews DW (1996) Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J. 15: 4130–4141.
Article CAS PubMed PubMed Central Google Scholar - Lee ST, Hoeflich KP, Wasfy GW, Woodgett JR, Leber B, Andrews DW, Hedley DW and Penn LZ (1999) Bcl-2 targeted to the endoplasmic reticulum can inhibit apoptosis induced by Myc but not etoposide in Rat-1 fibroblasts. Oncogene 18: 3520–3528.
Article CAS PubMed Google Scholar - McCullough KD, Martindale JL, Klotz LO, Aw TY and Holbrook NJ (2001) Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell Biol. 21: 1249–1259.
Article CAS PubMed PubMed Central Google Scholar - Nicotera P and Orrenius S (1998) The role of calcium in apoptosis. Cell Calcium 23: 173–180.
Article CAS PubMed Google Scholar - Budd SL and Nicholls DG (1996) Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurochem. 67: 2282–2291.
Article CAS PubMed Google Scholar - Rizzuto R and Pozzan T (2006) Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol. Rev. 86: 369–408.
Article CAS PubMed Google Scholar - Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M and Reed JC (1997) Channel formation by antiapoptotic protein Bcl-2. Proc. Natl. Acad. Sci. USA 94: 5113–5118.
Article CAS PubMed PubMed Central Google Scholar - Minn AJ, Velez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M and Thompson CB (1997) Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature 385: 353–357.
Article CAS PubMed Google Scholar - Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL and Distelhorst CW (1994) Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc. Natl. Acad. Sci. USA 91: 6569–6573.
Article CAS PubMed PubMed Central Google Scholar - Baffy G, Miyashita T, Williamson JR and Reed JC (1993) Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3-dependent hematopoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J. Biol. Chem. 268: 6511–6519.
CAS PubMed Google Scholar - Distelhorst CW and Shore GC (2004) Bcl-2 and calcium: controversy beneath the surface. Oncogene 23: 2875–2880.
Article CAS PubMed Google Scholar - Duchen MR (2000) Mitochondria and calcium: from cell signalling to cell death. J. Physiol. 529 (Part 1): 57–68.
Article CAS PubMed PubMed Central Google Scholar - Rizzuto R, Simpson AW, Brini M and Pozzan T (1992) Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358: 325–327.
Article CAS PubMed Google Scholar - Llopis J, McCaffery JM, Miyawaki A, Farquhar MG and Tsien RY (1998) Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA 95: 6803–6808.
Article CAS PubMed PubMed Central Google Scholar - Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA and Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763–1766.
Article CAS PubMed Google Scholar - Hajnoczky G, Csordas G, Krishnamurthy R and Szalai G (2000) Mitochondrial calcium signaling driven by the IP3 receptor. J. Bioenerg. Biomembr. 32: 15–25.
Article CAS PubMed Google Scholar - Denton RM, McCormack JG and Edgell NJ (1980) Role of calcium ions in the regulation of intramitochondrial metabolism. Effects of Na+, Mg2+ and ruthenium red on the Ca2+-stimulated oxidation of oxoglutarate and on pyruvate dehydrogenase activity in intact rat heart mitochondria. Biochem. J. 190: 107–117.
Article CAS PubMed PubMed Central Google Scholar - McCormack JG and Denton RM (1990) The role of mitochondrial Ca2+ transport and matrix Ca2+ in signal transduction in mammalian tissues. Biochim. Biophys. Acta 1018: 287–291.
Article CAS PubMed Google Scholar - Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R and Thomas AP (1998) Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 17: 4987–5000.
Article CAS PubMed PubMed Central Google Scholar - Lasorsa FM, Pinton P, Palmieri L, Fiermonte G, Rizzuto R and Palmieri F (2003) Recombinant expression of the Ca(2+)-sensitive aspartate/glutamate carrier increases mitochondrial ATP production in agonist-stimulated Chinese hamster ovary cells. J. Biol. Chem. 278: 38686–38692.
Article CAS PubMed Google Scholar - Hajnoczky G, Robb-Gaspers LD, Seitz MB and Thomas AP (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82: 415–424.
Article CAS PubMed Google Scholar - Jouaville LS, Pinton P, Bastianutto C, Rutter GA and Rizzuto R (1999) Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 96: 13807–13812.
Article CAS PubMed PubMed Central Google Scholar - Szalai G, Krishnamurthy R and Hajnoczky G (1999) Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 18: 6349–6361.
Article CAS PubMed PubMed Central Google Scholar - Di Lisa F and Bernardi P (2005) Mitochondrial function and myocardial aging. A critical analysis of the role of permeability transition. Cardiovasc. Res. 66: 222–232.
Article CAS PubMed Google Scholar - Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, Bernardi P and Bonaldo P (2003) Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat. Genet. 35: 367–371.
Article CAS PubMed Google Scholar - Rudolf R, Mongillo M, Rizzuto R and Pozzan T (2003) Looking forward to seeing calcium. Nat. Rev. Mol. Cell. Biol. 4: 579–586.
Article CAS PubMed Google Scholar - Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T and Rizzuto R (2000) Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J. Cell. Biol. 148: 857–862.
Article CAS PubMed PubMed Central Google Scholar - Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP, Demaurex N and Krause KH (2000) Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 97: 5723–5728.
Article CAS PubMed PubMed Central Google Scholar - Palmer AE, Jin C, Reed JC and Tsien RY (2004) Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. USA 101: 17404–17409.
Article CAS PubMed PubMed Central Google Scholar - Chiesa A, Rapizzi E, Tosello V, Pinton P, de Virgilio M, Fogarty KE and Rizzuto R (2001) Recombinant aequorin and green fluorescent protein as valuable tools in the study of cell signalling. Biochem. J. 355: 1–12.
Article CAS PubMed PubMed Central Google Scholar - Wang NS, Unkila MT, Reineks EZ and Distelhorst CW (2001) Transient expression of wild-type or mitochondrially targeted Bcl-2 induces apoptosis, whereas transient expression of endoplasmic reticulum-targeted Bcl-2 is protective against Bax-induced cell death. J. Biol. Chem. 276: 44117–44128.
Article CAS PubMed Google Scholar - Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T and Rizzuto R (2001) The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 20: 2690–2701.
Article CAS PubMed PubMed Central Google Scholar - Zhang J, Alter N, Reed JC, Borner C, Obeid LM and Hannun YA (1996) Bcl-2 interrupts the ceramide-mediated pathway of cell death. Proc. Natl. Acad. Sci. USA 93: 5325–5328.
Article CAS PubMed PubMed Central Google Scholar - Rippo MR, Malisan F, Ravagnan L, Tomassini B, Condo I, Costantini P, Susin SA, Rufini A, Todaro M, Kroemer G and Testi R (2000) GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J. 14: 2047–2054.
Article CAS PubMed Google Scholar - Chami M, Gozuacik D, Saigo K, Capiod T, Falson P, Lecoeur H, Urashima T, Beckmann J, Gougeon ML, Claret M, le Maire M, Brechot C and Paterlini-Brechot P (2000) Hepatitis B virus-related insertional mutagenesis implicates SERCA1 gene in the control of apoptosis. Oncogene 19: 2877–2886.
Article CAS PubMed Google Scholar - Ma TS, Mann DL, Lee JH and Gallinghouse GJ (1999) SR compartment calcium and cell apoptosis in SERCA overexpression. Cell Calcium 26: 25–36.
Article CAS PubMed Google Scholar - Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T and Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139.
Article CAS PubMed Google Scholar - Chami M, Prandini A, Campanella M, Pinton P, Szabadkai G, Reed JC and Rizzuto R (2004) Bcl-2 and Bax exert opposing effects on Ca2+ signalling, which do not depend on their putative pore-forming region. J. Biol. Chem. 279: 54581–54589.
Article CAS PubMed Google Scholar - Putney Jr JW (1990) Capacitative calcium entry revisited. Cell Calcium 11: 611–624.
Article CAS PubMed Google Scholar - Hofer AM, Fasolato C and Pozzan T (1998) Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: a study using simultaneous measurements of ICRAC and intraluminal [Ca2+]. J. Cell. Biol. 140: 325–334.
Article CAS PubMed PubMed Central Google Scholar - Vanden Abeele F, Skryma R, Shuba Y, Van Coppenolle F, Slomianny C, Roudbaraki M, Mauroy B, Wuytack F and Prevarskaya N (2002) Bcl-2-dependent modulation of Ca(2+) homeostasis and store-operated channels in prostate cancer cells. Cancer Cell. 1: 169–179.
Article CAS PubMed Google Scholar - Vanoverberghe K, Vanden Abeele F, Mariot P, Lepage G, Roudbaraki M, Bonnal JL, Mauroy B, Shuba Y, Skryma R and Prevarskaya N (2004) Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell. Death Differ. 11: 321–330.
Article CAS PubMed Google Scholar - Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P and Distelhorst CW (2004) Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J. Cell. Biol. 166: 193–203.
Article CAS PubMed PubMed Central Google Scholar - Distelhorst CW, Lam M and McCormick TS (1996) Bcl-2 inhibits hydrogen peroxide-induced ER Ca2+ pool depletion. Oncogene 12: 2051–2055.
CAS PubMed Google Scholar - He H, Lam M, McCormick TS and Distelhorst CW (1997) Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J. Cell. Biol. 138: 1219–1228.
Article CAS PubMed PubMed Central Google Scholar - Zhong F, Davis MC, McColl KS and Distelhorst CW (2006) Bcl-2 differentially regulates Ca2+ signals according to the strength of T cell receptor activation. J. Cell. Biol. 172: 127–137.
Article CAS PubMed PubMed Central Google Scholar - Ichimiya M, Chang SH, Liu H, Berezesky IK, Trump BF and Amstad PA (1998) Effect of Bcl-2 on oxidant-induced cell death and intracellular Ca2+ mobilization. Am. J. Physiol. 275: C832–C839.
Article CAS PubMed Google Scholar - Wei H, Wei W, Bredesen DE and Perry DC (1998) Bcl-2 protects against apoptosis in neuronal cell line caused by thapsigargin-induced depletion of intracellular calcium stores. J. Neurochem. 70: 2305–2314.
Article CAS PubMed Google Scholar - Kuo TH, Kim HR, Zhu L, Yu Y, Lin HM and Tsang W (1998) Modulation of endoplasmic reticulum calcium pump by Bcl-2. Oncogene 17: 1903–1910.
Article CAS PubMed Google Scholar - Breckenridge DG, Stojanovic M, Marcellus RC and Shore GC (2003) Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell. Biol. 160: 1115–1127.
Article CAS PubMed PubMed Central Google Scholar - Bastianutto C, Clementi E, Codazzi F, Podini P, De Giorgi F, Rizzuto R, Meldolesi J and Pozzan T (1995) Overexpression of calreticulin increases the Ca2+ capacity of rapidly exchanging Ca2+ stores and reveals aspects of their lumenal microenvironment and function. J. Cell. Biol. 130: 847–855.
Article CAS PubMed Google Scholar - Xu XZ, Moebius F, Gill DL and Montell C (2001) Regulation of melastatin, a TRP-related protein, through interaction with a cytoplasmic isoform. Proc. Natl. Acad. Sci. USA 98: 10692–10697.
Article CAS PubMed PubMed Central Google Scholar - Pinton P, Ferrari D, Rapizzi E, Di Virgilio FD, Pozzan T and Rizzuto R (2001) The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 20: 2690–2701.
Article CAS PubMed PubMed Central Google Scholar - Nakamura K, Bossy-Wetzel E, Burns K, Fadel MP, Lozyk M, Goping IS, Opas M, Bleackley RC, Green DR and Michalak M (2000) Changes in endoplasmic reticulum luminal environment affect cell sensitivity to apoptosis. J. Cell. Biol. 150: 731–740.
Article CAS PubMed PubMed Central Google Scholar - Campanella M, de Jong AS, Lanke KW, Melchers WJ, Willems PH, Pinton P, Rizzuto R and van Kuppeveld FJ (2004) The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J. Biol. Chem. 279: 18440–18450.
Article CAS PubMed Google Scholar - Chami M, Ferrari D, Nicotera P, Paterlini-Brechot P and Rizzuto R (2003) Caspase-dependent alterations of Ca2+ signaling in the induction of apoptosis by hepatitis B virus X protein. J. Biol. Chem. 278: 31745–31755.
Article CAS PubMed Google Scholar - Schwab BL, Guerini D, Didszun C, Bano D, Ferrando-May E, Fava E, Tam J, Xu D, Xanthoudakis S, Nicholson DW, Carafoli E and Nicotera P (2002) Cleavage of plasma membrane calcium pumps by caspases: a link between apoptosis and necrosis. Cell. Death Differ. 9: 818–831.
Article CAS PubMed Google Scholar - Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E and Nicotera P (2005) Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 120: 275–285.
Article CAS PubMed Google Scholar - Bian X, Hughes Jr FM, Huang Y, Cidlowski JA and Putney Jr JW (1997) Roles of cytoplasmic Ca2+ and intracellular Ca2+ stores in induction and suppression of apoptosis in S49 cells. Am. J. Physiol. 272: C1241–C1249.
Article CAS PubMed Google Scholar - Pinton P, Ferrari D, Di Virgilio F, Pozzan T and Rizzuto R (2001) Molecular machinery and signalling events in apoptosis. Drug Dev. Res. 52: 558–570.
Article CAS Google Scholar - Berridge MJ, Bootman MD and Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell. Biol. 4: 517–529.
Article CAS PubMed Google Scholar - Halestrap AP, Doran E, Gillespie JP and O'Toole A (2000) Mitochondria and cell death. Biochem. Soc. Trans. 28: 170–177.
Article CAS PubMed Google Scholar - Petronilli V, Penzo D, Scorrano L, Bernardi P and Di Lisa F (2001) The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 276: 12030–12034.
Article CAS PubMed Google Scholar - Assefa Z, Bultynck G, Szlufcik K, Nadif KN, Vermassen E, Goris J, Missiaen L, Callewaert G, Parys JB and De Smedt H (2004) Caspase-3-induced truncation of type 1 inositol trisphosphate receptor accelerates apoptotic cell death and induces inositol trisphosphate-independent calcium release during apoptosis. J. Biol. Chem. 279: 43227–43236.
Article CAS PubMed Google Scholar - Nakayama T, Hattori M, Uchida K, Nakamura T, Tateishi Y, Bannai H, Iwai M, Michikawa T, Inoue T and Mikoshiba K (2004) The regulatory domain of the inositol 1,4,5-trisphosphate receptor is necessary to keep the channel domain closed: possible physiological significance of specific cleavage by caspase 3. Biochem. J. 377: 299–307.
Article CAS PubMed PubMed Central Google Scholar - Schendel SL, Montal M and Reed JC (1998) Bcl-2 family proteins as ion-channels. Cell. Death Differ. 5: 372–380.
Article CAS PubMed Google Scholar - Matsuyama S, Schendel SL, Xie Z and Reed JC (1998) Cytoprotection by Bcl-2 requires the pore-forming alpha5 and alpha6 helices. J. Biol. Chem. 273: 30995–31001.
Article CAS PubMed Google Scholar - Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T and Korsmeyer SJ (2005) Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 102: 105–110.
CAS PubMed Google Scholar - White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB and Foskett JK (2005) The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat. Cell. Biol. 7: 1021–1028.
Article CAS PubMed PubMed Central Google Scholar - Hanson CJ, Bootman MD and Roderick HL (2004) Cell signalling: IP3 receptors channel calcium into cell death. Curr. Biol. 14: R933–R935.
Article CAS PubMed Google Scholar
Acknowledgements
This work was supported by Telethon Grant GGP05284, the Italian Association for Cancer Research (AIRC), the Italian University Ministry (MIUR and FIRB), the EU (fondi strutturali Obiettivo 2), the PRRIITT program of the Emilia Romagna Region and the Italian Space Agency (ASI).
Author information
Authors and Affiliations
- Department of Experimental and Diagnostic Medicine, Section of General Pathology, ER-GenTech laboratory and Interdisciplinary Center for the Study of Inflammation (ICSI), University of Ferrara, Via Borsari 46, Ferrara, I-44100, Italy
P Pinton & R Rizzuto
Corresponding author
Correspondence toR Rizzuto.
Additional information
Edited by C Borner
Rights and permissions
About this article
Cite this article
Pinton, P., Rizzuto, R. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum.Cell Death Differ 13, 1409–1418 (2006). https://doi.org/10.1038/sj.cdd.4401960
- Received: 06 March 2006
- Revised: 05 April 2006
- Accepted: 06 April 2006
- Published: 26 May 2006
- Issue Date: 01 August 2006
- DOI: https://doi.org/10.1038/sj.cdd.4401960