MicroRNAs in addiction: adaptation’s middlemen? (original) (raw)
. Author manuscript; available in PMC: 2014 Dec 2.
Published in final edited form as: Mol Psychiatry. 2011 May 24;16(12):1159–1168. doi: 10.1038/mp.2011.58
Abstract
A central question in addiction is how drug-induced changes in synaptic signaling are converted into long-term neuroadaptations. Emerging evidence reveals that microRNAs (miRNAs) have a distinct role in this process through rapid response to cellular signals and dynamic regulation of local mRNA transcripts. Because each miRNA can target hundreds of mRNAs, relative changes in the expression of miRNAs can greatly impact cellular responsiveness, synaptic plasticity and transcriptional events. These diverse consequences of miRNA action occur through coordination with genes implicated in addictions, the most compelling of these being the neurotrophin BDNF, the transcription factor cAMP-responsive element-binding protein (CREB) and the DNA-binding methyl CpG binding protein 2 (MeCP2). In this study, we review the recent progress in the understanding of miRNAs in general mechanisms of plasticity and neuroadaptation and then focus on specific examples of miRNA regulation in the context of addiction. We conclude that miRNA-mediated gene regulation is a conserved means of converting environmental signals into neuronal response, which holds significant implications for addiction and other psychiatric illnesses.
Keywords: addictions, mechanisms, miRNA, plasticity, regulation
Introduction
The past decade has seen a significant shift in the conceptualization of the genome. Non-coding elements, once considered ‘junk sequences’ (that is, evolutionary relics), are now known to be important regulators of gene expression necessary for the development and organization of complex life.1 This new framework allows extensive novel investigation into non-coding RNA, as 98% of the human genome is non-protein coding.2 One such class of non-coding RNAs, called microRNAs (miRNAs), are short (22 nucleotides), evolutionarily conserved regulatory molecules that directly target the 3′-untranslated region (3′-UTR) of mRNAs to modulate gene expression post-transcriptionally.3 miRNAs are endogenous to mammalian cells4 and are essential controllers of cellular proliferation, differentiation and apoptosis.5,6 In humans, more than 1000 miRNA sequences have thus far been identified.7 More than a third of all genes are subject to miRNA regulation, with each miRNA family targeting an average of ~500 RNA transcripts.8
Several properties of miRNA regulation and processing make them ideally suited for rapid environmental response. First, their small size and non-coding nature allow them to be transcribed more quickly than other immediate-early response genes, which are much longer and must undergo the additional step of translation.9 Second, because miRNAs target mRNAs directly, they regulate protein synthesis at the ribosome. Furthermore, their association with the ribosome allows subcellular localization, including to dendrites.9,10 The localization of miRNAs and their processing machinery to dendritic compartments provide a means for altered gene regulation in direct response to synaptic activity, fulfilling a unique requirement of neurons for synapse-specific adaptation as distinct from cell-wide changes in gene expression.11
Given their ubiquitous nature as well as the enrichment of many miRNAs in the brain,12,13 it is not surprising that they have been implicated in an ever-increasing number of neurological diseases. During the past several years, miRNA involvement has been associated with central nervous system disorders such as Tourette’s syndrome14 and Rett syndrome;15 neurodegenerative diseases such as Parkinson’s,16 Huntington’s,17 and Alzheimer’s disease18,19 and psychiatric disorders such as schizophrenia20,21 and addiction.22,23 An important role for miRNAs in addiction is supported by their established role in synaptic plasticity. Long-term facilitation, wherein neuronal synapses alter in strength according to activity at the synapse, is regarded as the underlying mechanism of addiction, compulsion and dependence,24-26 and drugs of abuse alter synaptic signaling in various brain regions, particularly the ventral tegmental area,27 striatum,28 nucleus accumbens29 and prefrontal cortex.30 miRNAs, by their modes of expression and action, are thus uniquely equipped to respond to altered synaptic signaling and induce neuroadaptation.
miRNAs in synaptic plasticity and neuronal regulation
The extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) family is a class of signal-transducing enzymes activated by cell-surface receptors and chemical or physical stresses.31 MAPK/ERK signaling regulates local miRNA expression via phosphorylation of the miRNA-generating complex,32 providing a common means through which an extracellular signal can be rapidly converted to an miRNA-mediated response. There are two general types of adaptive response under miRNA regulatory control: direct regulation of protein synthesis by miRNAs has a crucial role in plasticity at the synapse,33,34 whereas interactions of miRNAs with transcription factors seem to modulate more enduring neuroplastic changes on the level of the entire cell.
Dendritic morphology
Dendritic miRNAs may underlie or enhance the observed effects of key molecules in synaptic plasticity. Brain-derived neurotrophic factor (BDNF) is a neurotrophin crucial to cortical survival and maintenance, as well as the growth of new neurons and synapses.35,36 BDNF can induce transcription of miRNA-containing gene loci37 and interact directly with mature miRNAs.38 Treatment of neonatal rat cortical cells with BDNF upregulates the miRNA precursor premiR-132 through its action on cAMP-response element binding protein (CREB).39 In turn, mature miR-132 stimulates neurite outgrowth (the process preceding synapse formation) through inhibition of p250GAP, a protein that represses neurogenesis.39 Increased thickness of dendritic spines from transgenic miR-132 has been confirmed by an in vivo study.40 Another miRNA-mediated role for BDNF has been found in post-natal rat hippocampal cells: the brain-specific miR-134, located in the synaptodendritic compartment, inhibits translation of co-localized Lim domain-containing protein kinase 1.38 Lim domain-containing protein kinase 1 is a regulator of actin filament dynamics, necessary for dendritic spine development.41 Treatment with BDNF releases miR-134’s inhibition of Lim domain-containing protein kinase 1, thereby stimulating growth of the dendritic spine. This mechanism is reversible, with the activity state of the synapse switching translational inhibition on or off,38 illustrating a dynamic role for this miRNA in synapse plasticity.
Two recent studies42,43 provide additional evidence of localized miRNAs affecting dendritic structure. In the first, miR-138 was shown to restrict dendritic growth in rat hippocampal neurons through inhibition of acyl protein thioesterase 1.42 Calcium influx decreased pre-miR-138 levels and miR-138 cleavage activity, facilitating dendritic strengthening in response to synaptic stimulation. In a study of fragile-X mental retardation protein-related miRNAs in mouse hippocampal neurons, overexpression of either miR-132 or miR-125b led to opposing dendrite morphologies, with miR-132 corresponding to thicker spines and miR-125b to thinner.43 Interestingly, knockout of fragile-X mental retardation protein prevented these effects despite the lack of an miRNA recognition site, indicating an indirect association that regulates downstream protein targets in tandem.43 Taken together, these findings provide evidence that localization of miRNAs to synapses and their subsequent regulation of protein synthesis in response to specific synaptic stimuli is a significant mechanism underlying plasticity.
Gene regulation and memory
miRNA targeting of transcription factors such as CREB provides an additional layer of regulation with more widespread and enduring neuronal consequences. CREB-induced transcription is an important component of a switch from short-term to long-term plasticity,44 and proper CREB functioning is necessary for long-term memory formation.45 Heightened CREB concentrations increase neuronal excitability in the amygdala and nucleus accumbens,46,47 and the degree of CREB phosphorylation has been associated with sensitization to cocaine48 and morphine.49
Comparative sequence analysis reveals that miR-NAs expressed in neurons are highly enriched with cAMP-response elements and neuron-restrictive-silencing elements, implicating CREB as a positive regulator and RE1-silencing transcription factor (REST) as a common repressor of these genes.50 Because some neuronal miRNAs also target CREB and REST, and because all three types of regulators share neural gene targets, it is proposed that a network among CREB, REST and miRNAs carries out coordinated gene regulation through extensive feedback. Modeled gene networks indicate that feedback circuits increase the stability and robustness of the system,51 and mutual binding sites and targets shared by miRNAs and transcription factors could be seen as enabling cross-talk between the levels of genome, transcriptome and proteome.
The most abundant miRNA in the brain, miR-124,52 shows mutual targeting with REST. This antagonistic relation is important to cellular differentiation and identity through opposing effects on neural and non-neural transcripts,53 with miR-124 promoting a neuronal phenotype.54 In mature neurons, miR-124 also inhibits CREB in an activity-dependent manner.55 miR-124 responds to the neurotransmitter serotonin (5-HT) in Aplysia neurons with a rapid decrease in expression, leading to an increase in CREB expression and inducing long-term facilitation.55 Under normal conditions, this effect required five spaced pulses of 5-HT; however, when miR-124 was downregulated or CREB was upregulated, long-term facilitation could be induced after a single pulse. Thus, miR-124 and CREB can be seen to work in conjunction to mediate neural responsiveness to serotonin-induced learning.
Gao and colleague56 identified another miRNA–CREB pathway important in memory. Deficiency of SIRT1 was found to impair plasticity and memory formation in mice and cause overexpression of miR-134, an miRNA previously implicated in dendritic morphology. miR-134 was predicted to have three binding sites in the 3′-UTR of CREB mRNA, and luciferase reporter assays confirmed direct binding. It was found that SIRT1 normally forms an inhibitory complex upstream of the miR-134 gene to regulate its expression negatively. When disinhibited, overexpression of miR-134 downregulates CREB and BDNF (which is CREB activated) and leads to impaired plasticity and memory deficits. Blocking miR-134 in SIRT1-knockout mice was able to reverse these deficits.56 Intriguingly, Renthal et al.57 reported that chronic, but not acute, cocaine exposure increases the expression of SIRT1 in the nucleus accumbens, and that SIRT1 inhibition decreases the rewarding effects of the drug. Cocaine’s upregulation of SIRT1 could conceivably exert the observed effects via the miR-NA–CREB pathway discussed.
Chromatin remodeling may represent an additional component of a coordinated mechanism with miRNAs and transcription factors through which signaling-induced neuroadaptations gain long-term stability. miR-132, the CREB-activated miRNA involved in dendrite morphogenesis, also orchestrates chromatin remodeling through regulation of MeCP2, p300 and JARID1A in the suprachiasmatic nucleus, with the effect of attenuated resetting of the circadian clock in response to light.58 MeCP2 is a DNA-binding protein that can compact chromatin structure,59 repress transcription by competitive binding at promoters or through complex formation with histone deacetylases or co-repressors,60 or activate transcription through association with CREB1.61 MeCP2 is abundantly expressed in neurons and is critical to proper functioning; its over- and under-expression both result in detrimental neural effects, and mutations in the MeCP2 underlie Rett syndrome.62 There is recent evidence that MeCP2 regulates a cohort of miRNAs (including miR-132) through binding at promoter regions of miRNA transcription units, where it acts primarily as a repressor.63 Several of these miRNAs were found to be synaptically enriched, and many were also predicted to target BDNF, which is downregulated in MeCP2-knockout mouse and rescues Rett syndrome-like deficits.64 In turn, miR-132 represses MeCP2 but is activated by BDNF, highlighting an miRNA autoregulatory loop,63 which apparently stabilizes activity-dependent BDNF production, as well as MeCP2 expression.65
miR-132, CREB, MeCP2 and BDNF have all been demonstrated to be important components of learning and memory.40,45,66,67 An emerging picture is that these distinct molecular entities form a multi-level network that responds to neural activity at the immediate level of protein functioning at the synapse, but also, uses continuing feedback at the transcriptional and post-transcriptional level to carry out longer-term changes necessary for memory formation. The precise relations of such complex epigenetic networks are yet to be elucidated—particularly at the level of the miRNA, where the vast number of potential targets presents a practical challenge. However, miRNAs seem to occupy a unique position between synaptic signaling and neuronal gene expression, which holds significant consequences for memory as well as addiction.
miRNAs in substance use disorders
As drug addictions are widely regarded as disorders of plasticity, according to reward-based learning,24 it could be expected that miRNA-mediated mechanisms of synaptic plasticity such as those just described in functional systems contribute to formation of the addictive phenotype. During the past few years, evidence has begun to accumulate that miRNA responses to drug-induced stimuli have important roles in neuroadaptive pathways that are induced by, or react against, consistent drug exposure.
Cocaine addiction
A recent study by Hollander and colleagues68 reported altered miRNA expression in the striatum, a brain region involved in drug-seeking habits.69 Increased expression of miR-132 and the closely related miR-212 were observed in rats having extended access (6h per day), but not in rats under restricted access or ‘yoked’ rats, who received cocaine in a response-independent manner. Increasing or decreasing miR-212 expression was found to decrease and increase cocaine self-administration under unlimited access, indicating that this miRNA decreases cocaine’s motivational properties and protects against over-consumption. miR-212 appears to exert this effect, at least in part, by upregulating striatal CREB.68 Upregulation of the cAMP pathway is a compensatory response to chronic drug exposure,70 and elevation of CREB in the nucleus accumbens decreases the rewarding effects of cocaine.71 Follow-up work from the same research group72 demonstrated that MeCP2 forms a homeostatic interaction with miR-212 to control BDNF expression and cocaine intake. MeCP2 attenuates cocaine’s upregulation of miR-212 and subsequent CREB signaling, whereas miR-212 inhibits MeCP2 expression. Although there is evidence that MeCP2 itself acts as a transcriptional repressor of BDNF in the absence of neuronal activity,62 MeCP2 levels coordinate closely with BDNF levels in the brain,73 and phosphorylation of MeCP2 regulates activity-dependent expression of BDNF.74 Together, these observations indicate that a BDNF-MeCP2-inclusive network such as that described previously is necessarily co-expressed in response to neural activity and is engaged by cocaine. BDNF expression in the nucleus accumbens produces robust behavioral consequences, facilitating compulsive cocaine-taking behavior and increasing measured cocaine reward.75,76 As CREB induces both BDNF77 and MeCP265 expression, it seems that miR-212, by suppressing MeCP2 (and subsequently BDNF), serves as a ‘filter’ for CREB-responsive genes.72
The upregulation of miR-212 in the striatum may reflect a mechanism of tolerance within a neuron. Each use of cocaine upregulates BDNF,78 and BDNF action on its TrkB receptor is one of several types of synaptic activity that induces transcription of miR-212 and miR-132,37 which accounts for the observed increase in miR-212 in rats having extended access to cocaine. A sustained increase would reduce normative activity-dependent BDNF expression, which would decrease the rewarding effects of each cocaine exposure. More cocaine would therefore be necessary to achieve the same effect (Figure 1).
Figure 1.
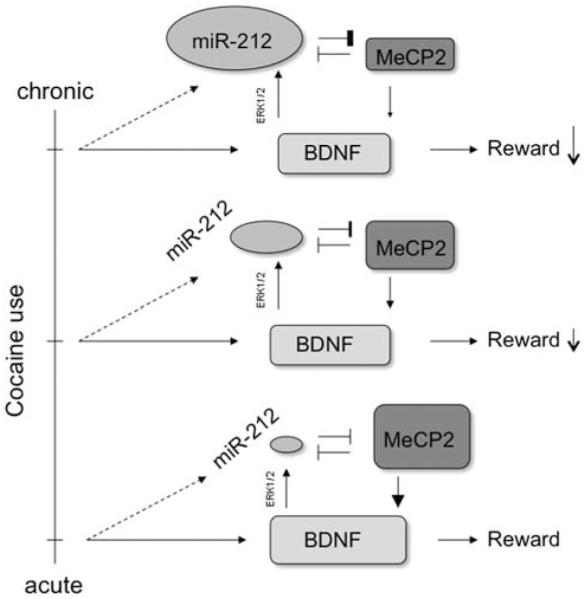
Relation between miR-212, methyl CpG-binding protein (MeCP2), and brain-derived neurotrophic factor (BDNF) mediates the adaptive response to chronic cocaine exposure. Cocaine increases BDNF concentrations, even after a single dose,78 and BDNF has a strong role in the motivating and rewarding aspects of the drug.86 BDNF signaling at the synapse increases transcription of miR-212 via an extracellular-signal-related kinase (ERK1/2) pathway.37 This miRNA exhibits mutual inhibition with MeCP2, a transcription factor necessary for BDNF expression in response to neural activity.74 The increased expression of miR-212 observed after chronic cocaine treatment72 therefore represents a mechanism of tolerance by inhibiting activity-dependent BDNF transcription in the nucleus accumbens.
Chandrasekar and Dreyer79 applied miRNA prediction software to identify miRNAs that might target cocaine-responsive genes implicated in addiction and found strong prediction for miR-124, let-7d and miR-181a. miRNA quantification of rat mesolimbic brain slices showed that miR-124 and let-7d were significantly downregulated and miR-181a was significantly upregulated by chronic cocaine administration. Further investigation80 revealed that overexpression of miR-124 and let-7d in the nucleus accumbens attenuates cocaine-induced conditioned place preference, whereas miR-181a overexpression enhanced cocaine-induced conditioned place preference, and silencing of these miRNAs produced inverse effects. This study further demonstrated an impressive array of addiction-related gene expression changes in these various conditions. Notably, miR-124 and let-7d overexpression upregulated the dopamine transporter, whereas miR-181a overexpression downregulated it. Because dopamine transporter is cocaine’s directly inhibited target and the source of its effects on the dopaminergic system,81 these findings likely relate strongly to the observed effects of manipulation of these miRNAs on conditioned place preference, an indirect measure of cocaine reward, and reflect compensatory changes in the cases of miR-124 and let-7d, and a sensitizing change in the case of miR-181a. The expression of a number of other genes is modulated by these miRNAs, including ΔFos and Fos B, DRD2 and DRD3, Nac1, Per2, GRIA2 and 7MYT1, highlighting the diverse effects of miRNA dysregulation on synaptic signaling (via receptors) and transcription factors. In light of the networks examined in this report, the effects of these miRNA manipulations on BDNF, CREB and MeCP2 are of particular interest. BDNF expression was decreased when miR-124 was silenced, or when let-7d was either overexpressed or silenced; MeCP2 was significantly downregulated when any of the three miRNAs was silenced, with the strongest effect (a 10-fold decrease) in the case of miR-124. Amounts of CREB protein also increased significantly when miR-124 was silenced.81
Although the transcriptional repressor REST was not examined in the more recent investigation, the initial study by Chandrasekar and Dreyer79 found that chronic cocaine exposure induces REST expression. The previously discussed antagonistic relation between miR-124 and REST might reasonably imply that this is another instance of a homeostatic relation between an miRNA and a transcription factor as an adaptation to chronic cocaine exposure. miR-124 and REST both target and suppress BDNF, so it seems that REST induction might serve to transfer control of BDNF inhibition from the translational level (via miRNA regulation) to the transcriptional level (via inhibition by REST). This shift may be necessary to allow an adaptive increase in CREB that was observed in the miR-124 silencing condition, as CREB is targeted by miR-124 but not REST (Figure 2). An inverse transfer of BDNF regulatory control has been observed in the prefrontal cortex: BDNF mRNA has been found to correlate more strongly with mechanisms of transcriptional control (for example, open-chromatin-associated histone H3 methylation) during childhood, whereas miRNAs became more prominent regulators in adolescence and adulthood.82 As miR-124 promotes the neuronal phenotype whereas REST opposes it, the observed effects of cocaine on these factors might be postulated to represent a regression into a more ‘immature’ neuronal phenotype. Observed effects of chromatin remodeling of the BDNF gene in response to chronic but not acute cocaine treatment83,84 also support such a shift in control. Regardless, the combined effects on CREB are likely relevant to learning-related aspects of addiction, such as cocaine-induced cues,80 as well as general cocaine reward.71
Figure 2.
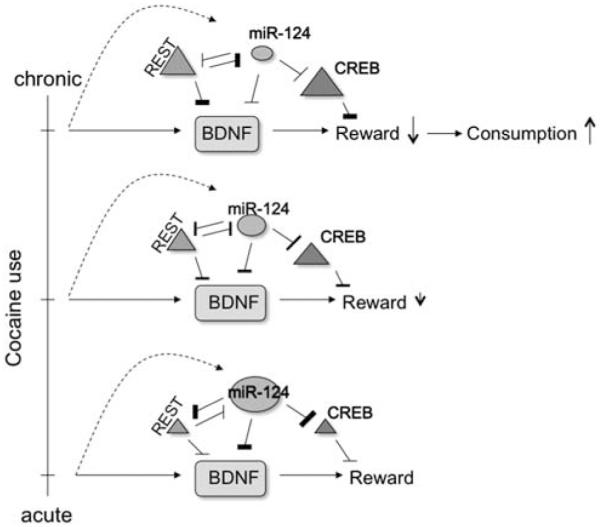
Decreased miR-124 expression in chronic cocaine conditions allows sustained increases in cAMP response element-binding protein (CREB). miR-124 is important in learning and memory through its inhibition of CREB.55 Following chronic cocaine exposure, this miRNA is down-regulated, whereas two of its targets, CREB and RE1-silencing transcription factor (REST), are upregulated.79 Both miR-124 and REST inhibit BDNF expression, so it seems that downregulation of this miRNA marks a shift in the control of BDNF inhibition from miR-124 to REST. This shift allows higher concentrations of CREB, which decreases the rewarding effects of cocaine.111
Whereas greater amounts of CREB protein in the nucleus accumbens decrease cocaine self-administration and relapse,85 more BDNF in this region increases self-administration and relapse.86 This is a somewhat surprising disparity, given that CREB induces BDNF transcription, but appears to be indicative of a general counter-balance between adaptations that accentuate cocaine’s signaling effects and those that offset these effects (even within the same network). miRNAs such as miR-124 and miR-212 appear to mediate this balance by selective targeting and activity-dependent expression.
Nicotine dependence
A characteristic shared feature of addictive drugs is an unconditional increase in synaptic dopamine,87 so dopamine receptor expression represents a potentially important factor in drug response. We recently investigated the differential expression of the dopamine receptor D1 gene (DRD1) in response to nicotine.23 This gene was previously found in a genetics association study to contain a single-nucleotide polymorphism (rs686) significantly associated with nicotine dependence (ND).88 Because the polymorphism rs686 was in the 3′-UTR, we hypothesized that such a significant genetic association of the polymorphism with ND might be mediated by miRNA. Investigation of candidate miRNAs revealed that mir-504 directly targeted DRD1, with the surprising effect of upregulating expression. Moreover, upregulation was significantly greater with the ‘A’ allele associated with ND. This observed effect agrees with the stronger predicted binding energy of miR-504 to the transcript containing this allele, as was confirmed by assay with an miR-504 inhibitor.23 The role of dopamine signaling in reward and motivation suggests that this miRNA-mediated pathway may underlie continued smoking behavior by increasing dopamine D1 receptor synthesis at nicotine-affected synapses. It may also affect plasticity downstream, as D1-receptor signaling phosphorylates CREB,89 and phosphorylated CREB in the nucleus accumbens is necessary for nicotine-induced conditioned place preference.90
In another study, we used an miRNA microarray approach to investigate the broad effects of nicotine stimulation on miRNA expression in rat PC12 cells.22 From several hundred probe sets, 25 miRNAs were found to show significant changes, evidence that nicotine exerts specific but widespread effects on miRNA regulation. One of these, miR-140*, showed a strong predicted binding site on dynamin1 (Dnm1), a large GTPase important for synaptic endocytosis that is significantly associated with ND.91 We subsequently demonstrated that this miRNA is greatly upregulated in response to nicotine treatment and binds directly to Dnm1 to inhibit its expression.22 Dynamin 1 may have a key role in chemical dependence through its action in signal termination of G-protein-coupled receptors, which include dopamine and opioid receptors, as changes in sensitivity of these receptors underlie acute drug effects.92 Moreover, morphine has been distinguished from non-addictive analogs by exhibiting a deficient ability to induce endocytosis of its receptor, which disrupts signal termination and desensitization.93 As dynamin 1 is crucial to endocytosis of G-protein-coupled receptors,94 its downregulation by miR-140* might contribute to the highly addictive aspects of nicotine, such as tolerance and craving.
Interestingly, several of the miRNAs found in this study to undergo nicotine-induced changes in expression also have been implicated in schizophrenia and other neurodegenerative disorders. For example, miR-181b, which is upregulated by nicotine, is upregulated in the temporal cortex of schizophrenic patients,95 whereas miR-30a-5p (one of the miRNAs that target BDNF in the prefrontal cortex82) and miR-29c are both downregulated by nicotine and are downregulated in the prefrontal cortex of postmortem brains in schizophrenia.20 As schizophrenia and ND show a strikingly high degree of co-morbidity,96,97 a more directed study of shared miRNA mechanisms in the two disorders could be very telling; it is possible that miRNAs could account for the co-morbidity, either through exacerbation of psychotic symptoms in response to drug use or by exerting similar effects in response to both anti-psychotics and cigarettes, thus fitting with a self-medication hypothesis.
miRNAs also appear to link nicotine with Alzheimer’s disease. miR-125b was found to be upregulated by nicotine and in the hippocampus of Alzheimer’s disease (AD) patients,98 whereas miR-93, upregulated by nicotine, is downregulated in the cortex in AD.99 Perhaps the most compelling correlate is miR-328, which is upregulated by nicotine and appears to have a significant role in the etiology of AD. Studies using postmortem tissues have revealed higher concentrations of β-amyloid precursor protein-converting enzyme protein (BACE1) in the brains of AD patients,100,101 which leads to the build-up of β-amyloid, a major component of senile plaques etiologic of AD, thought to be responsible for neurodegeneration.102 BACE1 is a predicted target of miR-328, and in a rodent model of AD, this miRNA was found to target and suppress BACE1 expression.103 Because nicotinic receptor stimulation protects neurons against β-amyloid toxicity,102 it is tempting to speculate that miR-328 upregulation by nicotine may be a component of the pathway underlying this protective effect. Regardless, the appearance of nicotine-responsive miRNAs in the etiology of neuropsychiatric disorders generally supports a role for these regulators in neural functioning. Table 1 provides a list of miRNAs dysregulated both in response to drugs and in neuropsychiatric illness.
Table 1.
miRNAs responsive to drugs of abuse that have also been associated with neuropsychiatric disorders
| miRNA | Targeted gene(s) | Biological function(s) | Associated diseases |
|---|---|---|---|
| mir-124 | REST53 CREB55 BDNF79 | Neuronal identity53 5-HT-induced learning55 Plasticity79 | Cocaine addiction,79 Alzheimer’s disease17 |
| miR-132 | P250GAP39 | Neurogenesis39 | Cocaine addiction,68 Huntington’s disease17 |
| miR-181b | VSNL195 GLIA195 | Intracellular signaling95 Neurotransmission95 | Nicotine dependence,22 schizophrenia95 |
| miR-30a-5p miR-29c | BDNF82 | Plasticity | |
| miR-125b | Lin-2817 NR2A43 | Neurogenesis17 Neurotransmission/plasticity43 | Nicotine dependence,22 Alzheimer’s disease17 |
| miR-93 miR-328 | VEGF112 BACE1103 | Cellular signaling112 Axon guidance, potentiation113 | ND,22 Alzheimer’s disease,17 schizophrenia113 |
Alcoholism
Alcohol exposure induces differential expression of about 2% of miRNAs in murine liver.104 Many of these miRNAs are also expressed in the brain, so it will be important to see whether alcohol exerts similar effects in this context.33 In murine striatal neurons and in adult rat neurons, miR-9 undergoes significant upregulation in response to alcohol and appears to contribute to alcohol tolerance through its regulation of the BK channel.105 This channel is highly relevant to neuronal function, as it regulates excitability, shaping of action potentials and neurotransmitter release.33,106 In mammals, alcohol evokes tolerance of BK channels.107 Intriguingly, miR-9 preferentially targets and degrades transcripts of BK channel isoforms sensitive to alcohol potentiation, whereas transcripts encoding alcohol-tolerant channels tend to lack miR-9-binding sites in their 3′-UTRs.33 Thus, alcohol-induced upregulation of miR-9 shifts BK channel expression toward more tolerant isoforms. miR-9 also targets DRD2,33 and lower expression of this receptor has been associated with alcohol abuse,108 indicating that this miR-9 might influence the rewarding effect of alcohol in addition to its involvement in tolerance.
A systems genetic analysis of alcohol consumption has found that variations underlying GABAergic brain function contribute a significant genetic component, and that G protein subunit beta 1 (Gnβ1) represents a candidate transcript for miRNA regulation relevant to alcohol consumption, based on differential 3′-UTR sequences (and predicted binding affinities) between various intensities of alcohol consumption.109 The strongest target predictions across multiple software platforms were for miR-101a/b and miR-218. Subsequent studies to investigate the actual effects of these miRNAs on alcohol consumption will be necessary for confirmation. In support of a role for regulation of the GABAergic system in alcoholism, infusion of a GABAA alpha siRNA vector (pHSVsiLA2) into the central nucleus caused a reduction of binge drinking in alcohol-preferring rats.110 This study represents a promising implementation of gene therapy given the successful behavioral effect and the tight control of the micro-infusion to specific brain regions.110
Perspective
From initial drug exposure to chemical dependence and addiction, there is a panoply of molecular changes that comprise neural adaptation. Recent studies on the effects of drugs of abuse on miRNAs reveal that these tiny regulatory molecules can have either a contributing role in the development of addiction, as in the case of miR-504 increasing DRD1 expression, or a counteractive role against drug stimulatory effects, as in the case of miR-212 upregulating CREB. These converse biological effects represent the ‘pull’ and ‘push’ of addiction: the response of motivation-based learning networks toward perceived reward vs. the counter-response of neuronal homeostasis against sustained alterations in extracellular signaling. Although these effects of sensitization and tolerance are divergent, both appear to be multi-level adaptations, spanning from short-term changes in signaling cascades to long-term changes in baseline gene expression. miRNAs can respond to synaptic signals (for example, miR-124’s response to 5-HT) and regulate local protein synthesis, but also mediate transcription factors and chromatin remodelers; thus we propose that they are uniquely suited for neuroadaptation by converting short-term into long-term plasticity.
Plasticity relies on coordinated changes among vastly complex molecular networks, and drugs of abuse seem to exert their effect not via a single member of the network, but through coordination. However, one conserved mechanism among these gene networks appears to be miRNA-constrained feedback loops, wherein a drug-induced stimulus acts as an impetus for a change in gene expression through a temporary effect on an miRNA or transcription factor before a balance is restored through feedback. Particularly among activity-dependent species, such as BDNF, CREB and MeCP2, in which precise spatiotemporal regulation is essential, feedback loops would be necessary for both stability and efficiency during complex, associative changes within the molecule. Furthermore, the observations of miRNA-CREB interactions in mediating neuronal responsiveness might suggest a role for miRNAs as markers of recent neural activity, thus providing a context for subsequent network activation. A more precise investigation of the temporally dependent effects of neuronal (and particularly hippocampal) miRNA expression compared with early activation genes might further elucidate this question.
A fuller characterization of miRNA species and their targets will be crucial to a fuller understanding of this type of gene regulation and to practical application. Deep sequencing studies are already allowing large-scale profiles of miRNA populations, but it also will be necessary to characterize differential miRNA expression among different cell types and at specific synapses to fully understand their functional roles. Given the growing specificity of our knowledge ofmiRNA targets, their ability to modulate numerous downstream targets makes them attractive as potential therapeutic targets, because such manipulations might affect an entire network rather than a single species. In addiction especially, the potential to inhibit the longer-term adaptations to drugs of abuse would be helpful in stopping the progression of the disorder and decreasing relapse risk. Of course, the enduring risks of such gene manipulations must be addressed more thoroughly in pre-clinical trials than we have yet seen.
Although miRNAs appear uniquely situated to participate in cross-talk between cellular signaling and long-term gene expression, the precise means and the degree of specificity of such a phenomenon is a mystery. Within a neuron, the relative level of expression of a gene, for example BDNF, will certainly affect its chromatin structure, in essence because the ’supply’ (transcription) of the gene must match the demand. Is it possible that the corresponding levels of the gene’s targeting miRNA, for example, miR-132, might also be factored into the gene’s chromatin remodeling? In this example, the answer seems to be yes, through miR-132’s regulation of MeCP2, which in turn regulates BDNF. The tantalizing possibility, however, is that something similar is happening on a much larger scale, perhaps using miRNAs combinatorially.
Although the full power of miRNAs as gene regulators remains to be seen, they certainly seem to have a significant role in proper brain functioning. Their adaptive nature is distinctly suited for a role in addiction, but their dysregulation is also being observed increasingly in schizophrenia, Parkinson’s and Alzheimer’s. As our ability to understand gene networks increases in both scope and precision, we will certainly want to be attentive to these tiny regulatory molecules, as the early evidence suggests they may serve as critical links.
Acknowledgments
The preparation of this report was in part supported by NIH Grants DA-12844 and DA-13787 to Ming D Li. We thank Dr David L Bronson for his excellent editing of this report.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Perkins DO, Jeffries C, Sullivan P. Expanding the ‘central dogma’: the regulatory role of nonprotein coding genes and implications for the genetic liability to schizophrenia. Mol Psychiatry. 2005;10:69–78. doi: 10.1038/sj.mp.4001577. [DOI] [PubMed] [Google Scholar]
- 2.Mattick JS. Non-coding RNAs: the architects of eukaryotic complexity. Embo Reports. 2001;2:986–991. doi: 10.1093/embo-reports/kve230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 4.Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 5.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 6.Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer. 2006;94:776–780. doi: 10.1038/sj.bjc.6603023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun JG, Liao RX, Qiu J, Jin JY, Wang XX, Duan YZ, et al. Microarray-based analysis of microRNA expression in breast cancer stem cells. J Exp Clin Canc Res. 2010;29:174. doi: 10.1186/1756-9966-29-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman RC, Farh KKH, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hobert O. Gene regulation by transcription factors and microRNAs. Science. 2008;319:1785–1786. doi: 10.1126/science.1151651. [DOI] [PubMed] [Google Scholar]
- 10.Ashraf SI, McLoon AL, Sclarsic SM, Kunes S. Synaptic protein synthesis associated with memory is regulated by the RISC pathway in Drosophila. Cell. 2006;124:191–205. doi: 10.1016/j.cell.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 11.Martin KC, Zukin RS. RNA trafficking and local protein synthesis in dendrites: an overview. J Neurosci. 2006;26:7131–7134. doi: 10.1523/JNEUROSCI.1801-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lugli G, Torvik VI, Larson J, Smalheiser NR. Expression of microRNAs and their precursors in synaptic fractions of adult mouse forebrain. J Neurochem. 2008;106:650–661. doi: 10.1111/j.1471-4159.2008.05413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abelson JF, Kwan KY, O’Roak BJ, Baek DY, Stillman AA, Morgan TM, et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science. 2005;310:317–320. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- 15.Urdinguio RG, Fernandez AF, Lopez-Nieva P, Rossi S, Huertas D, Kulis M, et al. Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics. 2010;5:656–663. doi: 10.4161/epi.5.7.13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, et al. A microRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317:1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maes OC, Chertkow HM, Wang E, Schipper HM. MicroRNA: implications for Alzheimer disease and other human CNS disorders. Curr Genomics. 2009;10:154–168. doi: 10.2174/138920209788185252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bicker S, Schratt G. microRNAs: tiny regulators of synapse function in development and disease. J Cell Mol Med. 2008;12:1466–1476. doi: 10.1111/j.1582-4934.2008.00400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sethi P, Lukiw WJ. Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci Lett. 2009;459:100–104. doi: 10.1016/j.neulet.2009.04.052. [DOI] [PubMed] [Google Scholar]
- 20.Perkins DO, Jeffries CD, Jarskog LF, Thomson JM, Woods K, Newman MA, et al. microRNA expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biol. 2007;8:R27. doi: 10.1186/gb-2007-8-2-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu YL, Kalbfleisch T, Brennan MD, Li Y. A microRNA gene is hosted in an intron of a schizophrenia-susceptibility gene. Schizophr Res. 2009;109:86–89. doi: 10.1016/j.schres.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang WH, Li MD. Nicotine modulates expression of miR-140*, which targets the 3′-untranslated region of dynamin 1 gene (Dnm1) Int J Neuropsychopharmacol. 2009;12:537–546. doi: 10.1017/S1461145708009528. [DOI] [PubMed] [Google Scholar]
- 23.Huang WH, Li MD. Differential allelic expression of dopamine D1 receptor gene (DRD1) is modulated by microRNA miR-504. Biol Psychiatry. 2009;65:702–705. doi: 10.1016/j.biopsych.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Ann Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- 25.Hyman SE, Malenka RC. Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci. 2001;2:695–703. doi: 10.1038/35094560. [DOI] [PubMed] [Google Scholar]
- 26.Koob GF. The neurocircuitry of addiction: implications for treatment. Clin Neurosci Res. 2005;5:89–101. [Google Scholar]
- 27.Kauer JA. Learning mechanisms in addiction: synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol. 2004;66:447–475. doi: 10.1146/annurev.physiol.66.032102.112534. [DOI] [PubMed] [Google Scholar]
- 28.Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM. It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 2003;26:184–192. doi: 10.1016/S0166-2236(03)00065-1. [DOI] [PubMed] [Google Scholar]
- 29.Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, Nestler EJ. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010;33:267–276. doi: 10.1016/j.tins.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalivas PW, Volkow N, Seamans J. Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron. 2005;45:647–650. doi: 10.1016/j.neuron.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Chang LF, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 32.Paroo Z, Ye XC, Chen S, Liu QH. Phosphorylation of the human microRNA-generating complex mediates MAPK/Erk signaling. Cell. 2009;139:112–122. doi: 10.1016/j.cell.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pietrzykowski AZ. The role of microRNAs in drug addiction: a big lesson from tiny molecules. Int Rev Neurobiol. 2010;91:1001–1005. doi: 10.1016/S0074-7742(10)91001-5. [DOI] [PubMed] [Google Scholar]
- 34.Smalheiser NR, Lugli G. microRNA regulation of synaptic plasticity. Neuromol Med. 2009;11:133–140. doi: 10.1007/s12017-009-8065-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Acheson A, Conover JC, Fandl JP, Dechiara TM, Russell M, Thadani A, et al. A Bdnf autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–453. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- 36.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Ann Rev of Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Remenyi J, Hunter CJ, Cole C, Ando H, Impey S, Monk CE, et al. Regulation of the miR-212/132 locus by MSK1 and CREB in response to neurotrophins. Biochem J. 2010;428:281–291. doi: 10.1042/BJ20100024. [DOI] [PubMed] [Google Scholar]
- 38.Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, et al. A brain-specific microRNA regulates dendritic spine development. Nature. 2006;439:283–289. doi: 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]
- 39.Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, et al. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci U S A. 2005;102:16426–16431. doi: 10.1073/pnas.0508448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hansen KF, Sakamoto K, Wayman GA, Impey S, Obrietan K. Transgenic miR132 alters neuronal spine density and impairs novel object recognition memory. Plos One. 2010;5:e15497. doi: 10.1371/journal.pone.0015497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Endo M, Ohashi K, Sasaki Y, Goshima Y, Niwa R, Uemura T, et al. Control of growth cone motility and morphology by LIM kinase and slingshot via phosphorylation and dephosphorylation of cofilin. J Neurosci. 2003;23:2527–2537. doi: 10.1523/JNEUROSCI.23-07-02527.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siegel G, Obernosterer G, Fiore R, Oehmen M, Bicker S, Christensen M, et al. A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat Cell Biol. 2009;11:705–U736. doi: 10.1038/ncb1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edbauer D, Neilson JR, Foster KA, Wang CF, Seeburg DP, Batterton MN, et al. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron. 2010;65:373–384. doi: 10.1016/j.neuron.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/s0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- 45.Benito E, Barco A. CREB’s control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33:230–240. doi: 10.1016/j.tins.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 46.Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, et al. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci. 2009;12:1438–1443. doi: 10.1038/nn.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, et al. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9:475–477. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- 48.Marin MT, Berkow A, Golden SA, Koya E, Planeta CS, Hope BT. Context-specific modulation of cocaine-induced locomotor sensitization and ERK and CREB phosphorylation in the rat nucleus accumbens. Eur J Neurosci. 2009;30:1931–1940. doi: 10.1111/j.1460-9568.2009.06982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moron JA, Gullapalli S, Taylor C, Gupta A, Gomes I, Devi LA. Modulation of opiate-related signaling molecules in morphine-dependent conditioned behavior: conditioned place preference to morphine induces CREB phosphorylation. Neuropsychophar-macology. 2010;35:955–966. doi: 10.1038/npp.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu J, Xie XH. Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol. 2006;7:R85. doi: 10.1186/gb-2006-7-9-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Becskei A, Serrano L. Engineering stability in gene networks by autoregulation. Nature. 2000;405:590–593. doi: 10.1038/35014651. [DOI] [PubMed] [Google Scholar]
- 52.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Current Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 53.Visvanathan J, Lee S, Lee B, Lee JW, Lee SK. The microRNA miR-124 antagonizes the anti-neural REST/SCP1 pathway during embryonic CNS development. Genes Dev. 2007;21:744–749. doi: 10.1101/gad.1519107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conaco C, Otto S, Han JJ, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad Sci U S A. 2006;103:2422–2427. doi: 10.1073/pnas.0511041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rajasethupathy P, Fiumara F, Sheridan R, Betel D, Puthanveettil SV, Russo JJ, et al. Characterization of small RNAs in Aplysia reveals a role for miR-124 in constraining synaptic plasticity through CREB. Neuron. 2009;63:803–817. doi: 10.1016/j.neuron.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao J, Wang WY, Mao YW, Graff J, Guan JS, Pan L, et al. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature. 2010;466:1105–1109. doi: 10.1038/nature09271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Renthal W, Kumar A, Xiao GH, Wilkinson M, Covington HE, Maze I, et al. Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron. 2009;62:335–348. doi: 10.1016/j.neuron.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alvarez-Saavedra M, Antoun G, Yanagiya A, Oliva-Hernandez R, Cornejo-Palma D, Perez-Iratxeta C, et al. miRNA-132 orchestrates chromatin remodeling and translational control of the circadian clock. Hum Mol Genet. 2011;20:731–751. doi: 10.1093/hmg/ddq519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Georgel PT, Horowitz-Scherer RA, Adkins N, Woodcock CL, Wade PA, Hansen JC. Chromatin compaction by human MeCP2. Assembly of novel secondary chromatin structures in the absence of DNA methylation. J Biol Chem. 2003;278:32181–32188. doi: 10.1074/jbc.M305308200. [DOI] [PubMed] [Google Scholar]
- 60.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 61.Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, et al. DNA methylation-related chromatin remodeling in activity-dependent Bdnf gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 63.Wu H, Tao JF, Chen PJ, Shahab A, Ge WH, Hart RP, et al. Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2010;107:18161–18166. doi: 10.1073/pnas.1005595107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Larimore JL, Chapleau CA, Kudo S, Theibert A, Percy AK, Pozzo-Miller L. Bdnf overexpression in hippocampal neurons prevents dendritic atrophy caused by Rett-associated MECP2 mutations. Neurobiol Dis. 2009;34:199–211. doi: 10.1016/j.nbd.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat Neurosci. 2007;10:1513–1514. doi: 10.1038/nn2010. [DOI] [PubMed] [Google Scholar]
- 66.Lonetti G, Angelucci A, Morando L, Boggio EM, Giustetto M, Pizzorusso T. Early environmental enrichment moderates the behavioral and synaptic phenotype of MeCP2 null mice. Biol Psychiatry. 2010;67:657–665. doi: 10.1016/j.biopsych.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 67.Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2010;107:22687–22692. doi: 10.1073/pnas.1012851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hollander JA, Im HI, Amelio AL, Kocerha J, Bali P, Lu Q, et al. Striatal microRNA controls cocaine intake through CREB signaling. Nature. 2010;466:197–202. doi: 10.1038/nature09202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Belin D, Everitt BJ. Cocaine seeking habits depend upon doparnine-dependent serial connectivity linking the ventral with the dorsal striatum. Neuron. 2008;57:432–441. doi: 10.1016/j.neuron.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 70.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 71.Carlezon WA, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, et al. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- 72.Im HI, Hollander JA, Bali P, Kenny PJ. MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat Neurosci. 2010;13:1120–1127. doi: 10.1038/nn.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 74.Zhou ZL, Hong EJ, Cohen S, Zhao WN, Ho HYH, Schmidt L, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schoenbaum G, Stalnaker TA, Shaham Y. A role for BDNF in cocaine reward and relapse. Nat Neurosci. 2007;10:935–936. doi: 10.1038/nn0807-935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Horger BA, Iyasere CA, Berhow MT, Messer CJ, Nestler EJ, Taylor JR. Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J Neurosci. 1999;19:4110–4122. doi: 10.1523/JNEUROSCI.19-10-04110.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Choi KH, Whisler K, Graham DL, Self DW. Antisense-induced reduction in nucleus accumbens cyclic AMP response element binding protein attenuates cocaine reinforcement. Neuroscience. 2006;137:373–383. doi: 10.1016/j.neuroscience.2005.10.049. [DOI] [PubMed] [Google Scholar]
- 78.Le Foll B, Diaz J, Sokoloff P. A single cocaine exposure increases BDNF and D-3 receptor expression: implications for drug-conditioning. Neuroreport. 2005;16:175–178. doi: 10.1097/00001756-200502080-00022. [DOI] [PubMed] [Google Scholar]
- 79.Chandrasekar V, Dreyer JL. microRNAs miR-124, let-7d and miR-181a regulate cocaine-induced plasticity. Mol Cell Neurosci. 2009;42:350–362. doi: 10.1016/j.mcn.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 80.Chandrasekar V, Dreyer JL. Regulation of MiR-124, Let-7d, and MiR-181a in the accumbens affects the expression, extinction, and reinstatement of cocaine-induced conditioned place preference. Neuropsychopharmacology. 2011;36:1149–1164. doi: 10.1038/npp.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mellios N, Huang HS, Grigorenko A, Rogaev E, Akbarian S. A set of differentially expressed miRNAs, including miR-30a-5p, act as post-transcriptional inhibitors of BDNF in prefrontal cortex. Hum Mol Genet. 2008;17:3030–3042. doi: 10.1093/hmg/ddn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DEH, Truong HT, et al. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 84.Sadri-Vakili G, Kumaresan V, Schmidt HD, Famous KR, Chawla P, Vassoler FM, et al. Cocaine-induced chromatin remodeling increases brain-derived neurotrophic factor transcription in the rat medial prefrontal cortex, which alters the reinforcing efficacy of cocaine. J Neurosci. 2010;30:11735–11744. doi: 10.1523/JNEUROSCI.2328-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Self DW, Genova LM, Hope BT, Barnhart WJ, Spencer JJ, Nestler EJ. Involvement of cAMP-dependent protein kinase in the nucleus accumbens in cocaine self-administration and relapse of cocaine-seeking behavior. J Neurosci. 1998;18:1848–1859. doi: 10.1523/JNEUROSCI.18-05-01848.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10:1029–1037. doi: 10.1038/nn1929. [DOI] [PubMed] [Google Scholar]
- 87.Hnasko TS, Sotak BN, Palmiter RD. Morphine reward in dopamine-deficient mice. Nature. 2005;438:854–857. doi: 10.1038/nature04172. [DOI] [PubMed] [Google Scholar]
- 88.Huang W, Ma JZ, Payne TJ, Beuten J, Dupont RT, Li MD. Significant association of DRD1 with nicotine dependence. Hum Genet. 2008;123:133–140. doi: 10.1007/s00439-007-0453-9. [DOI] [PubMed] [Google Scholar]
- 89.Xing B, Kong H, Meng X, Wei SG, Xu M, Li SB. Dopamine D1 but not D3 receptor is critical for spatial learning and related signaling in the hippocampus. Neurosci. 2010;169:1511–1519. doi: 10.1016/j.neuroscience.2010.06.034. [DOI] [PubMed] [Google Scholar]
- 90.Brunzell DH, Mineur YS, Neve RL, Picciotto MR. Nucleus accumbens CREB activity is necessary for nicotine conditioned place preference. Neuropsychopharmacology. 2009;34:1993–2001. doi: 10.1038/npp.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu Q, Huang WH, Payne TJ, Ma JZ, Li MD. Detection of genetic association and a functional polymorphism of dynamin 1 gene with nicotine dependence in European and African Americans. Neuropsychopharmacology. 2009;34:1351–1359. doi: 10.1038/npp.2008.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Koob GF, Nestler EJ. The neurobiology of drug addiction. J Neuropsychiatry Clin Neurosci. 1997;9:482–497. doi: 10.1176/jnp.9.3.482. [DOI] [PubMed] [Google Scholar]
- 93.Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- 94.Artalejo CR, Elhamdani A, Palfrey HC. Sustained stimulation shifts the mechanism of endocytosis from dynamin-1-dependent rapid endocytosis to clathrin- and dynamin-2-mediated slow endocytosis in chromaffin cells (vol 99, pg 6358, 2002) Proc Natl Acad Sci U S A. 2002;99:9082. doi: 10.1073/pnas.082658499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Beveridge NJ, Tooney PA, Carroll AP, Gardiner E, Bowden N, Scott RJ, et al. Dysregulation of miRNA 181b in the temporal cortex in schizophrenia. Hum Mol Genet. 2008;17:1156–1168. doi: 10.1093/hmg/ddn005. [DOI] [PubMed] [Google Scholar]
- 96.Volkow ND. Substance use disorders in schizophrenia - clinical implications of comorbidity. Schizophr Bull. 2009;35:469–472. doi: 10.1093/schbul/sbp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Williams JM, Gandhi KK, Lu SE, Kumar S, Shen JW, Foulds J, et al. Higher nicotine levels in schizophrenia compared with controls after smoking a single cigarette. Nicotine Tob Res. 2010;12:855–859. doi: 10.1093/ntr/ntq102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport. 2007;18:297–300. doi: 10.1097/WNR.0b013e3280148e8b. [DOI] [PubMed] [Google Scholar]
- 99.Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Holsinger RMD, McLean CA, Masters CL, Evin G, Beyreuther K. BACE and beta-secretase product CTF beta are increased in sporadic Alzheimer’s disease brain. Neurobiol Aging. 2002;23:S177. [Google Scholar]
- 101.Fukumoto H, Cheung B, Hyman B, Irizarry M. beta-site amyloid precursor protein cleaving enzyme (BACE) activity is increased in temporal neocortex of Alzheimer’s disease. Neurobiol Aging. 2002;23:S181. [Google Scholar]
- 102.Kihara T, Shimohama S, Sawada H, Kimura J, Kume T, Kochiyama H, et al. Nicotinic receptor stimulation protects neurons against beta-amyloid toxicity. Ann Neurol. 1997;42:159–163. doi: 10.1002/ana.410420205. [DOI] [PubMed] [Google Scholar]
- 103.Boissonneault V, Plante I, Rivest S, Provost P. MicroRNA-298 and MicroRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J Biol Chem. 2009;284:1971–1981. doi: 10.1074/jbc.M807530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dolganiuc A, Petrasek J, Kodys K, Catalano D, Mandrekar P, Velayudham A, et al. MicroRNA expression profile in Lieber-DeCarli diet-induced alcoholic and methionine choline deficient diet-induced nonalcoholic steatohepatitis models in mice. Alcohol Clin Exp Res. 2009;33:1704–1710. doi: 10.1111/j.1530-0277.2009.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pietrzykowski AZ, Friesen RM, Martin GE, Puig SI, Nowak CL, Wynne PM, et al. Posttranscriptional regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron. 2008;59:274–287. doi: 10.1016/j.neuron.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shipston MJ. Alternative splicing of potassium channels: a dynamic switch of cellular excitability. Trends Cell Biol. 2001;11:353–358. doi: 10.1016/s0962-8924(01)02068-2. [DOI] [PubMed] [Google Scholar]
- 107.Martin G, Puig SI, Pietrzykowski A, Zadek P, Emery P, Treistman S. Restricted cellular localization of a specific BK-channel subtype controls ethanol sensitivity in the nucleus accumbens. Alcohol Clin Exp Res. 2004;28:61A. doi: 10.1523/JNEUROSCI.0684-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Volkow ND, Wang GJ, Begleiter H, Porjesz B, Fowler JS, Telang F, et al. High levels of dopamine D-2 receptors in unaffected members of alcoholic families:possible protective factors. Arch Gen Psychiatry. 2006;63:999–1008. doi: 10.1001/archpsyc.63.9.999. [DOI] [PubMed] [Google Scholar]
- 109.Saba LM, Bennett B, Hoffman PL, Barcomb K, Ishii T, Kechris K, et al. A systems genetic analysis of alcohol drinking by mice, rats and men: influence of brain GABAergic transmission. Neuro-pharmacology. 2011;60:1269–1280. doi: 10.1016/j.neuropharm.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu J, Yang AR, Kelly T, Puche A, Esoga C, June HL, Jr, et al. Binge alcohol drinking is associated with GABAA alpha2-regulated Toll-like receptor 4 (TLR4) expression in the central amygdala. Proc Natl Acad Sci USA. 2011;108:4465–4470. doi: 10.1073/pnas.1019020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and Delta FosB. Nat Neurosci. 2003;6:1208–1215. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- 112.Long JY, Wang Y, Wang WJ, Chang BHJ, Danesh FR. Identification of microRNA-93 as a novel regulator of vascular endothelial growth factor in hyperglycemic conditions. J Biol Chem. 2010;285:23455–23463. doi: 10.1074/jbc.M110.136168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Santarelli DM, Beveridge NJ, Tooney PA, Cairns MJ. Upregulation of dicer and microRNA expression in the dorsolateral prefrontal cortex Brodmann area 46 in schizophrenia. Biol Psychiatry. 2011;69:180–187. doi: 10.1016/j.biopsych.2010.09.030. [DOI] [PubMed] [Google Scholar]