Tumour mutational burden: clinical utility, challenges and emerging improvements (original) (raw)
Introduction
Over the span of more than five decades of molecular cancer research and DNA sequencing, various genomic alterations within cancer cells have been identified and affirmed as the drivers of malignant transformation1,2. Tumour mutational burden (TMB) provides a measure to quantify the deviation of the cancer genome from its germline counterpart, encapsulated by the aggregate of somatic mutations. The TMB concept gained prominence following the conclusion of the initial large-scale cancer sequencing endeavours in 2013, accompanied by the observation of marked variations in TMB between and within different types of cancers3,4.
In this Review, we first discuss the clinical utility of TMB as a biomarker of benefit from immune-checkpoint inhibitors (ICIs). We then describe the different definitions of TMB that can be applied and summarize the underlying mutational processes. This is followed by discussions of the various factors influencing the measurement and analytical validity of TMB. After considering the clinical limitations of TMB, we explore emerging strategies that might improve the performance of this biomarker. These improvements can be grouped conceptually into refining (preselecting or weighing of the included mutations) and combining (developing a single, more-accurate biomarker by combining TMB with other molecular markers).
A biomarker for immuno-oncology
The proof of principle for TMB as a predictive biomarker in immuno-oncology was demonstrated by groundbreaking studies analysing cohorts of patients with advanced-stage melanoma receiving anti-CTLA4 antibodies and those with advanced-stage non-small-cell lung cancer (NSCLC) receiving anti-PD-1 antibodies in 2014 and 2015, respectively5,6. Building upon this foundation, the clinical adoption of TMB was propelled by two primary forces, both of which gained considerable clinical relevance around 2018. First, the introduction of ICIs as first-line therapies for patients with advanced-stage NSCLC, based on the results of Keynote-189 (ref. 7), Keynote-407 (ref. 8), Keynote-042 (ref. 9) and CheckMate-227 (ref. 10), together with similarly promising data from clinical trials involving patients with advanced-stage melanoma and renal cell carcinoma, as well as other cancer types, created the need for more-robust and complementary predictive biomarkers to refine patient stratification and to improve the allocation of therapeutic resources beyond tumour cell PD-L1 expression. Second, technological advances and decreasing costs rendered the use of next-generation sequencing (NGS) panels including 300–500 genes feasible in routine clinical practice, resulting in attempts to derive additional clinical utility from expanded tumour molecular profiling. Panel-based TMB thereby emerged as an elegant method of leveraging technological progress to address clinical needs and soon came to be regarded as a promising approach for the selection of patients who are most likely to benefit from ICIs.
Cancer immunotherapies exploit the presentation of tumour-specific antigens on the surface of cancer cells that can be recognized by the immune system (Fig. 1). In the first step of the pathway, the protein products of mutated genes are processed by cancer cells and presented as neoantigens but can also be picked up by professional antigen-presenting cells (APCs). APCs are then able to migrate to lymph nodes and present the antigens to T cell precursors, a process regulated by CTLA4. Finally, cancer cells presenting the same antigen can be recognized by primed cytotoxic T cells, a process regulated by PD-1 and its ligand PD-L1. Given that TMB reflects the quantity of antigens potentially available for processing, a higher TMB increases the probability of at least one highly immunogenic neoantigen being presented. The hypothesis of a higher efficacy of immunotherapies in patients with high TMB was investigated in various cohorts of patients receiving ICIs11. At the same time, the complexity of antigen processing, presentation and recognition, which can be defective in some patients, suggests that multiple other factors beyond TMB can contribute to the success of immunotherapies.
Fig. 1: Simplified representation of the effect of TMB on the tumour-specific immune response.
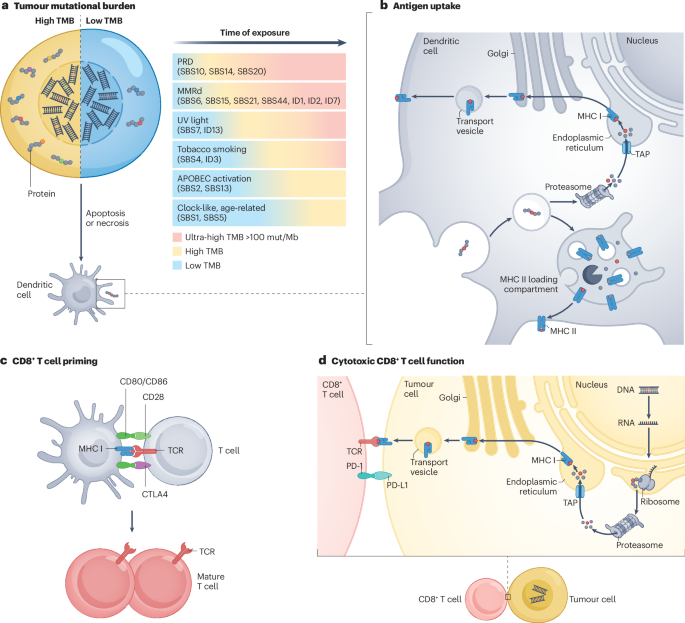
a, Tumour mutational burden (TMB), defined as the number of mutations present in the tumour genome, is the consequence of various processes that promote the accumulation of mutations, over varying time spans. The differing mutation rates associated with these processes determine the probability of a TMB-high phenotype. Defects in proofreading deficiency (PRD) and mismatch repair deficiency (MMRd) mechanisms are highly likely to result in a high or ultra-high TMB, whereas mutational processes associated with APOBEC activation or spontaneous deamination are less likely to do so. b, Antigen uptake by dendritic cells. Tumour-derived mutated proteins are internalized by professional antigen-presenting cells (APCs) such as dendritic cells and undergo intracellular processing, resulting in the generation of antigenic peptides. Neopeptides for presentation to cytotoxic (CD8+) T cells, which are crucial for T cell-mediated antitumour immunity, are loaded onto MHC class I molecules, whereas those intended for presentation to helper (CD4+) T cells are loaded onto MHC class II molecules. c, CD8+ T cell priming. After migration to lymph nodes, dendritic cells present processed tumour-derived neopeptides on MHC I molecules to naive CD8+ T cells (depicted in grey). The recognition of these antigen–MHC I complexes via the T cell receptor (TCR) is a prerequisite for T cell activation. A co-stimulatory signal provided by the interaction of CD28 with CD80/CD86 is also necessary for this activation. The primed T cells undergo clonal expansion and differentiate into effector T cells with specific functions, including cytotoxic T cells. This process is negatively regulated by CTLA4, which binds CD80/CD86 in an antagonistic manner to CD28 and promotes immune tolerance. d, Cytotoxic activity of CD8+ T cells. Activated CD8+ T cells recognize tumour-associated neopeptides presented by MHC I on the surface of tumour cells. This interaction triggers the release of cytotoxic molecules such as perforin and granzymes. Cancer cells might also express PD-L1, which interacts with PD-1 on CD8+ T cells, leading to suppression of T cell activity. The downstream signalling pathways activated by CTLA4 binding to CD80/CD86 as well as PD-L1 binding to PD-1 enable tumours to evade immune surveillance. Cancer cells with a high TMB are likely to produce greater numbers of neopeptides (part a) and are therefore more likely to have successful antigen uptake, processing and presentation (parts b and c) as well as an increased probability of recognition by mature (CD8+) T cells (part d). ID, small insertions and deletions signature; SBS, single-base substitution; TAP, transporter associated with antigen processing; UV, ultraviolet.
The high expectations surrounding the performance of TMB were fuelled by promising data from retrospective analyses of samples from some of the early trials testing ICIs. The phase III CheckMate-026 trial tested nivolumab versus platinum-based chemotherapy as first-line therapy for patients with stage IV PD-L1-positive NSCLC (≥1% on the 28-8 assay). In an exploratory analysis, TMB was assessed using whole-exome sequencing (WES) with TMB-high defined as being in the highest tertile for somatic missense mutations present in a baseline tumour sample (≥243). Comparisons of the outcomes of patients with TMB-high tumours in the nivolumab arm versus those with TMB-moderate or TMB-low tumours demonstrated higher objective response rates (ORRs; 47% versus 23%) and longer median progression-free survival (PFS; 9.7 months versus 3.6–4.2 months)12. Moreover, within the TMB-high subset, those who also had PD-L1-high (≥50%) tumours had an even more favourable ORR (75%), whereas PD-L1 expression had no predictive value in the TMB-low subset. Along the same lines, a post hoc analysis of data from the nivolumab plus ipilimumab arm of CheckMate-012 revealed a median PFS of 17.1 months for patients with TMB-high tumours (defined as having a TMB of above the median for the entire cohort) versus 3.7 months for their TMB-low counterparts. Patients with TMB-high and PD-L1-positive (≥1%) tumours again had the highest ORR (62.5%) with limited benefit in the TMB-low subgroup, regardless of PD-L1 expression13. These results suggest that TMB has predictive value complementary to that of PD-L1 expression and that the likelihood of deriving clinical benefit from ICIs is much higher for patients with TMB-high tumours than for those with TMB-low tumours. This notion was further corroborated by the analysis of PFS data from CheckMate-227 part 1, in which a high TMB was defined as ≥10 somatic mutations (coding base substitutions and short indels) per megabase (mut/Mb) according to the FoundationOne CDx assay. Patients with advanced-stage NSCLC who met this criterion derived significantly greater benefit from nivolumab plus ipilimumab compared with chemotherapy (7.2 months versus 5.5 months; HR 0.58, 97.5% CI 0.41–0.81; P < 0.001), regardless of PD-L1 expression14. Overall survival (OS) data from this trial demonstrated benefit from the ICI regimen compared with chemotherapy for patients with PD-L1-positive tumours (≥1%; 17.1 months versus 14.9 months; P = 0.007), which lead to the regulatory approval of this combination in this setting10. In a subgroup analysis of OS stratifying for both TMB and PD-L1, the highest level of benefit from nivolumab plus ipilimumab was reported for patients with TMB-high, PD-L1-low (<1%) tumours. These somewhat confusing results as well as possibly the approval of nivolumab plus ipilimumab with PD-L1, but not TMB as the companion biomarker, can be explained at least partly by the confounding study design of CheckMate-227 part 1: PFS in the TMB-high population (≥10 mut/Mb) but OS in the PD-L1 expressing population (≥1%) were selected as coprimary end points.
The association between high TMB and benefit from ICIs was subsequently validated across cancer types based on large amounts of retrospective and prospective data15,16,17,18,19. In a retrospective analysis of more than 3,000 patients with NSCLC, high TMB (defined as a harmonized TMB _z_-score ≥0) but not high PD-L1 levels was associated with responses to ICIs ≥24 months compared with short-term (<12-month) responses, demonstrating that TMB is the more-robust predictor of long-term benefit and a potential surrogate for this uncommon but profound clinical outcome20. An even greater level of enrichment of long-term responders could be achieved by applying the top 10% of TMB scores as the cut-off. Of note, PD-L1 expression did not positively correlate with TMB in any of these studies, indicating that the two biomarkers are largely independent both in patients with NSCLC and with other cancers21. Independence of the two biomarkers was confirmed by a lack of any significant correlation between PD-L1 mRNA levels and TMB in 21 cancer types profiled in The Cancer Genome Atlas (TCGA), albeit with the exception of microsatellite-unstable (MSI-H) and _POLE_-mutated tumours, which were characterized by simultaneous high PD-L1 mRNA levels and a high TMB22. These results support a role of TMB as a predictive biomarker of benefit from ICIs, independent of and additional to PD-L1.
The potential of TMB holds particular relevance for cancers of unknown primary (CUP), which account for ~3–5% of all cancers. These malignancies are distinguished by the unknown origin of the primary cancer cells and typically associated with a dire prognosis, with a 1-year survival of ~20% and median OS durations of ~3 months23. Data from several studies analysing the mutational profiles of CUPs demonstrate that ~10–20% of patients have a high TMB (applying cut-offs between 12 mut/Mb and 17 mut/Mb based on panel sequencing)24,25,26. In accordance with other tumour types27, in patients with relapsed and/or refractory CUP with a high (≥12 mut/Mb) TMB according to NGS with large panels, the combination of nivolumab plus ipilimumab leads to an ORR of 60% and a 1-year PFS of 60% compared with 7.7% and 9% in TMB-low patients, respectively28. Additionally, data from the randomized CUPISCO trial indicate that patients with newly diagnosed unfavourable subtype of CUP, as defined by the current guidelines23, and a TMB ≥16 mut/Mb have a significantly longer PFS when treated with atezolizumab when compared with standard chemotherapy29. By contrast, the addition of atezolizumab to platinum-based chemotherapy only marginally improved median PFS in previously untreated patients with CUP with a TMB <16 mut/Mb. Finally, data from a retrospective analysis of TMB in more than 8,000 patients across 24 cancer types confirm that 23.9% and 13.3% of CUPs have TMBs ≥10 mut/Mb and ≥20 mut/Mb, respectively, and that a significant association exists between TMB ≥ 10 mut/Mb and favourable OS on an anti-PD-1 or anti-PD-L1 antibody (HR 0.52, 95% CI 0.30–0.90), independent of microsatellite stability status30. The higher levels of benefit from ICIs among patients with TMB-high versus TMB-low CUP and data from the CUPISCO trial demonstrating an advantage of atezolizumab versus standard-of-care chemotherapy specifically in patients with TMB-high CUP support the clinical adoption of TMB as a biomarker in this setting.
Blood-based TMB (bTMB) offers a less-invasive alternative to tissue-based TMB that might have utility in settings in which obtaining sufficient-quality tumour tissue samples for molecular testing is challenging, including many patients with lung cancer, CUP, relapsed disease and metastases31,32,33. Data from the MYSTIC trial demonstrate the feasibility of TMB quantification from plasma samples and that a bTMB cut-off ≥20 mut/Mb is predictive of clinical benefit from durvalumab plus tremelimumab versus chemotherapy in patients with metastatic NSCLC34. Despite frequent assay failure (25–30%), for example, owing to samples with insufficient circulating tumour DNA (ctDNA), valid bTMB scores were obtained for 81% of patients compared with 63% for tissue-based TMB. Analysis of high-quality matched tumour and plasma samples revealed concordant results in 50% of patients, whereas 29% and 21% of mutations were detected only in tissue or blood samples, respectively. These findings are in line with the results of other similar studies involving patients with metastatic NSCLC32. Cohort C of the BFAST trial compared atezolizumab with chemotherapy as first-line therapy for patients with unresectable, advanced-stage NSCLC characterized by a bTMB cut-off of ≥10 mut/Mb, with the majority (291 of 472) having a bTMB ≥16 mut/Mb. Although the trial failed to achieve the primary end point of PFS in the bTMB ≥16 mut/Mb population (HR 0.77, 95% CI 0.59–1.00; P = 0.053), 18-month PFS and OS in this subgroup both favoured atezolizumab. Interestingly, when bTMB was evaluated using the F1L CDx assay with an equivalent cut-off of bTMB ≥13.60 mut/Mb in an exploratory analysis, atezolizumab significantly improved PFS versus chemotherapy (4.9 months versus 4.2 months, HR 0.71, 95% CI 0.52–0.97; P = 0.029)35. Further research should aim to address the utility of bTMB primarily in patients with advanced-stage disease, in whom obtaining sufficient tissue for molecular testing can be difficult, whereas ctDNA levels are more likely to be sufficient for NGS compared with earlier stage tumours shedding less DNA to the bloodstream.
The milestone achievement for TMB occurred when pembrolizumab received Accelerated Approval by the FDA for use across adult or paediatric patients with any TMB-high (≥10 mut/Mb) unresectable and/or advanced-stage solid tumours that have progressed following prior treatment and who have no satisfactory alternative treatment options available. This decision was based on outcomes of the single-arm Keynote-158 trial and followed an earlier similar approval with MSI-H as the biomarker. Despite the groundbreaking nature of this tumour-agnostic approval, this decision sparked several controversial discussions around the globe, culminating in a rejection of the approval application by the EMA36,37. Criticisms of Keynote-158 have three main directions36. First, this trial did not include a control arm and analysis of survival end points was lacking; only response rates were analysed at the time of FDA approval. Thus, randomized trials designed to demonstrate that ICIs improve survival outcomes in unselected patients with TMB-high tumours continue to be warranted. Second, sample sizes for most specific cancer types were low, particularly for cancer types that do not already have histology-specific approvals of ICIs. Third, the cut-off of 10 mut/Mb specified in the FDA approval needs further validation, as higher cutpoints are associated with gradual improvements in response rates.
Theoretical background
WES and sequencing with large panels (≥1 Mb) are the most common approaches for TMB measurement, the former typically based on sequencing of matched tumour and nonmalignant DNA and the latter typically based on tumour-only sequencing (Box 1). When calculated based on WES data, TMB can be measured directly and reported as the total number of missense mutations. Unlike panel-based sequencing, this method directly measures TMB without subsampling of the investigated sequence and thus without introducing a stochastic error. Alternatively, analysis of panel-based sequencing data provides a TMB reported as mut/Mb of the sequenced region, resulting in a demand for comparisons with WES and between panels of varying sizes. When panel sequencing is used, often additional types of non-synonymous mutations beyond missense mutations are included in the TMB calculation to enhance the total number of mutations investigated and thus reduce the stochastic error. At the same time, known oncogenic variants are excluded from the calculation to prevent TMB overestimation originating from the biased design of cancer-specific sequencing panels (which are enriched for oncogenes and tumour suppressor genes). A bridging study involving a cohort of patients with NSCLC demonstrated that a missense mutational burden of 199 mutations detected by WES corresponded to a TMB, including all exonic mutations, of 10 mut/Mb detected using the panel-based F1 CDx assay38. In this study, the overall level of agreement in classifying TMB as low or high was 84%. Such converting factors can also be extrapolated for other commercial and academic sequencing panels39,40.
Missense mutations are the most abundant type of mutation present in the exomes of cancer cells, with synonymous mutations, nonsense mutations and indels comprising 40%, 10% and 5% of the average missense mutational burden, respectively, although these proportions vary substantially both within and between cancer types41. Only non-synonymous mutations, which result in alterations of the amino acid sequence of a protein, contribute to the neoantigen burden, arguing for the exclusion of synonymous mutations from TMB. Furthermore, although rarer, indels are generally much more immunogenic than missense mutations42,43, suggesting that indel and missense mutational burden might be worth investigating either as separate metrics or in combination as part of a weighted metric.
TMB includes any somatic mutations accumulated before the onset of cancer as well as in numerous cancer cell clones evolving over time. In cancers originating from highly self-renewing tissues, such as chronic lymphocytic leukaemia and colorectal and ovarian cancers, more than half of the somatic mutations are estimated to have occurred before tumour initiation44. Specific aetiological factors and mechanisms of pathogenesis can be operative during different phases of tumour evolution (Fig. 1a). As a consequence, TMB is highly variable both within and between different cancer types3,45. To illustrate the variability of TMB within and across cancer types, we plotted missense mutation and indel burden for each tumour type included in the TCGA cohort (Fig. 2). The scatterplot demonstrates an association between high TMB and mutations associated with deficient DNA repair (primarily occurring in colorectal, gastric and endometrial cancers) as well as with exposure to tobacco smoke (mainly lung cancers) and UV light (melanoma). Tumours with mutations conferring proofreading deficiencies (PRDs) have an extremely high missense mutational burden (median 895, 2.5–97.5% quantile 25–11,100) and a high median indel burden (12, 0–429). Tumours with mismatch repair deficiency (MMRd) have a very high missense mutational burden (543, 112–9,000) and extremely high indel burden (82, 0–445). By comparison, missense mutational burden and indel burden were low in tumours with homologous recombination repair deficiencies (90, 23–707 and 5, 0–27) and even lower in DNA repair-proficient tumours (37, 4–474 and 2, 0–16). High TMBs are especially likely to be observed in tumours arising from exposure to potent mutagens, such as tobacco smoke and UV light, as seen in smoking-related lung cancer and cutaneous melanoma. Extremely high TMBs can also be observed in tumours with MMRd and PRD, a category mostly comprising adenocarcinomas of the colon, rectum, stomach and endometrium.
Fig. 2: Pan-cancer analysis of missense mutation and indel burden.
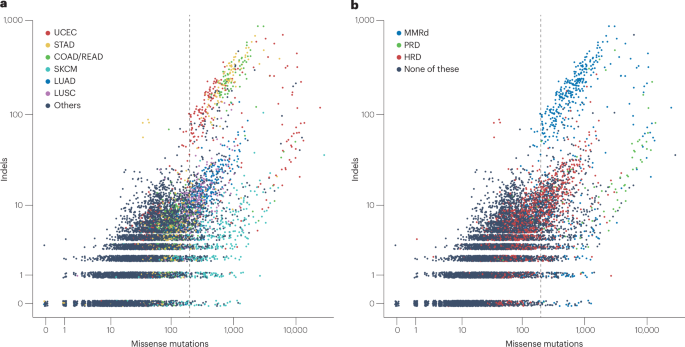
This analysis was performed using publicly available data from The Cancer Genome Atlas cohort[168](/articles/s41571-024-00932-9#ref-CR168 "The US National Cancer Institute Pan-Cancer Atlas. https://gdc.cancer.gov/about-data/publications/pancanatlas
(2024)."). Vertical lines mark the cut-off of 199 missense mutations, which separates TMB-high from TMB-low tumours, when analysed using whole-exome sequencing data. **a**, Missense mutation and indel burden classified by cancer types. **b**, Tumours classified by type of DNA repair deficiency. Mismatch repair deficiency (MMRd) was identified by analysing the proportion of signal mutations C(C > A)N, G(C > T)N and Y(A > G)N in tumours with at least 100 missense mutations, as described in detail elsewhere[169](/articles/s41571-024-00932-9#ref-CR169 "Maruvka, Y. E. et al. Analysis of somatic microsatellite indels identifies driver events in human tumors. Nat. Biotechnol. 35, 951–959 (2017)."). Proofreading deficiency (PRD) was determined by the detection of deleterious mutations in _POLE_ or _POLD1_. Homologous recombination repair deficiency (HRD) was determined based on an HRDsum score of at least 42\. COAD/READ, colorectal adenocarcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; UCEC, uterine corpus endometrial carcinoma.Measurement and confounders
The TMB of a tumour can be determined using WES of paired tumour and germline samples obtained from the same patient. Alternatively, TMB can be approximated using targeted panel sequencing approaches that enable assessments of only parts of the coding DNA sequence (CDS). Panel sequencing of tumour material only is usually performed in routine clinical practice, owing to simpler logistics and the avoidance of the efforts and costs of additional germline sequencing. By contrast, the addition of germline sequencing requires the implementation of parallel workflows, and the related logistics, for blood or nonmalignant tissue samples to secure turnaround within a clinically relevant time frame. In the tumour-only setting, germline mutations need to be filtered out using SNP databases (for common SNPs) and bioinformatic algorithms (for rare and/or private SNPs) instead of somatic variant calling, which is reliant on a baseline defined by a nonmalignant sample.
In addition to different laboratory assays, different definitions of TMB and different bioinformatic algorithms for its determination can also be used (Box 1). For example, definitions including only missense mutations have been used in many WES studies, whereas a broader range of mutation classes are included in the TMB calculation for most of the panel-based sequencing approaches. The differing laboratory and bioinformatic approaches used raise the important question of interassay comparability and call for efforts to bridge any gaps in performance between the different approaches. The harmonization of TMB measurements has been addressed in studies conducted by the Friends of Cancer Research and the German Quality in Pathology (QuIP) initiative39,40,46. Following comparisons of TMB results obtained using different assays and at different laboratories, the following general conclusions can be drawn: best laboratory and bioinformatic practices for TMB measurement should be defined in consensus guidelines; and universal reference standards should be used for TMB calibration to ensure optimal interassay and interlaboratory reproducibility of TMB cut-offs and the accuracy of any corresponding patient stratification. Data published in 2023 indicate that good levels of interlaboratory reproducibility are achievable (Pearson _R_-values of 0.97–0.99) for TMB measured using WES across five centres in Germany47.
The main potential confounders of TMB measurement include biological variability, technical variability, insufficient tumour purity, inexact filtering of germline variants when only tumour tissue is sequenced and stochastic variability when panel sequencing is performed. Several studies have investigated the biological variability of TMB by analysing two or more tissue blocks obtained from the same tumour. For samples from patients with NSCLC, concordant TMB classification (high versus low at a cut-off of 10 mut/Mb) across all tissue blocks was observed across 85–90% of tumours41,48,49.
Technical variability refers to differences in TMB that arise following repeat analysis of the same DNA sample. The extent of interassay and interlaboratory technical variations in TMB was investigated in the American Friends of Cancer Research and the German QuIP TMB studies39,40. Both laboratory-related and bioinformatics-related factors are crucial for successful and accurate measurements of TMB. Mutation calling needs to be sensitive but at the same time specific to separate mutations from possible sequencing artefacts that, for example, can occur as a consequence of formalin fixation. Investigators in the QuIP study identified the following factors as being key for successful TMB measurements: implementation of a read deduplication strategy to separate reads originating from different DNA strands from PCR duplicates, obtaining samples with sufficient tumour purity and sufficient amounts of DNA40.
Variant allele frequency (VAF) is defined as the ratio of the number of reads supporting a variant and the total number of reads aligned to the investigated sequence locus. In cancer tissues, VAF is influenced by the tumour purity of the sample, the clonality of the mutation and the number of affected and unaffected gene copies. For variant calling, a VAF threshold needs to be set to ensure sensitive and specific variant detection. In clinical practice, the VAF threshold is often set to 5%, so that for a sequencing coverage of 200×, all called variants are supported by at least 10 reads. Only variants above the VAF threshold are included in the TMB calculation.
Assessments of the implications of tumour purity for TMB measurements indicate significant positive correlations in samples from patients with lung adenocarcinoma, lung squamous cell carcinoma, urothelial carcinoma and other cancer types50. With these observations in mind, computer simulations were used to simulate the influence of tumour purity on the ability to detect TMB-high tumours (Fig. 3). Lower tumour purity leads to lower estimations of VAF and to a lower number of variants being deemed to contribute to the TMB. Therefore, we simulated lower tumour purity considering the copy number of the tumour genome at the position of the mutation and subsequently assessed whether the variant signal remained above the VAF threshold for inclusion in TMB (here 5%). In general, applying a simulated reduction in tumour purity resulted in lower VAFs, which in turn resulted in lower TMB. The median TMBs were reduced by 2%, 10% and 32% when tumour purities of 60%, 40% and 20%, respectively, were simulated and compared with a reference of 80%. For the 10% purity tumour samples with the poorest TMB estimation, TMB was reduced by 12%, 35% and 77%, or more. Relating these results to a cut-off of 199 mutations, the sensitivity for detection of high TMB in the pan-cancer cohort was 98%, 90% and 62% for tumour purities of 60%, 40% and 20%, respectively. For samples from patients with head and neck cancer, lung adenocarcinoma, lung squamous cell carcinoma and cutaneous melanoma with tumour purities of 40%, the sensitivity to detect high TMB was 80%, 89%, 92% and 91%, respectively. In summary, TMB measurement is accurate for samples with tumour purities of at least 60%, resulting in a sensitivity for the detection of a high TMB that is close to 100%. By contrast, TMB is substantially underestimated for tumour purities <40%, resulting in a reduced sensitivity and underdiagnosis of a substantial proportion of patients that might derive benefit from ICIs. Thus, the selection of tissue samples for TMB measurement should aim for a minimum tumour purity of 60% and definitely not <40%. The development of bioinformatic methods of accurately correcting TMB readings for low tumour purity is a potential method of mitigating the underdetection of TMB-high tumours when sample purity is low. Such a method has been developed and the corrected TMB generated using this approach outperforms classical TMB in the prediction of clinical outcome when applied retrospectively to cohorts of patients who received ICIs50. Other potential ways of improving the handling of low-purity tumour samples include increasing the sensitivity for mutation calling by increasing the sequencing depth or using clonal TMB (cTMB), a metric that includes only truncal mutations of a high VAF, as a potentially more-robust alternative to total TMB.
Fig. 3: Influence of tumour purity on TMB measurement.
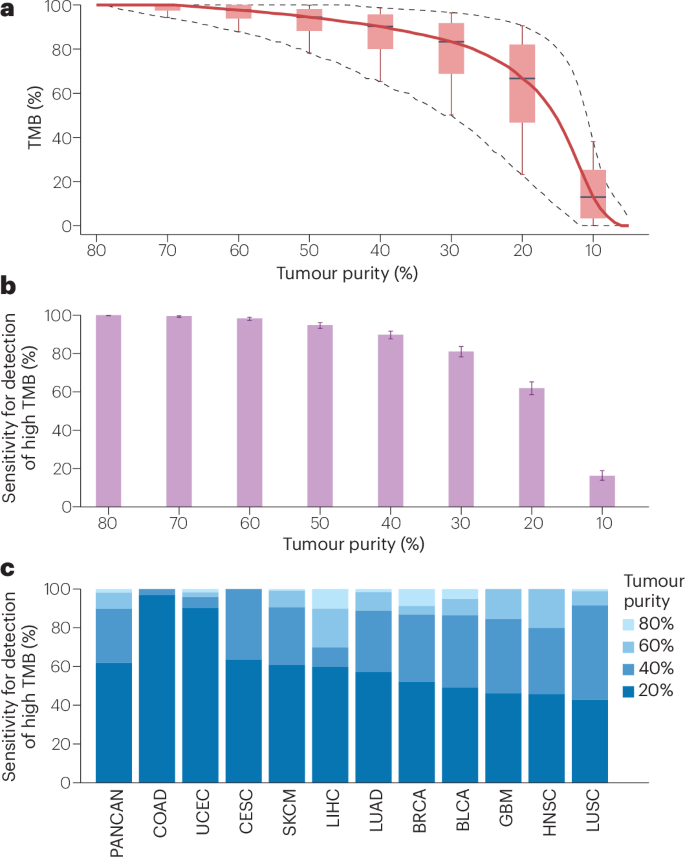
Simulation of different levels of tumour purity performed using publicly available data from The Cancer Genome Atlas (TCGA) subcohort of tumours with a purity ≥80% and available copy number data from genotyping (n = 4,900)[168](/articles/s41571-024-00932-9#ref-CR168 "The US National Cancer Institute Pan-Cancer Atlas. https://gdc.cancer.gov/about-data/publications/pancanatlas
(2024)."). We simulated cohorts with homogeneous tumour purities ranging from 10% to 80%. Tumour purities of 10–70% were compared with a tumour purity of 80%, which served as the reference. The tumour mutational burden (TMB) of a tumour with a simulated purity of _q_ was calculated as the number of missense mutations with a variant allele frequency (VAF) purity = _q_ ≥ 5%, in which the VAF of a variant of the simulated tumour is connected with that of the original by VAF (purity = _q_) = d(_p_)/d(_q_) × VAF (purity = _p_), with d(_x_) = 1 + 2/CN × (1/_x_ – 1). Herein, CN is the copy number of the tumour in the region of the mutation. **a**, TMB (in %) detected in tumours of lower purity relative to the reference. Boxes indicate the 10%, 25%, 50%, 75% and 90% quantiles. **b**, Sensitivity for the detection of TMB-high (≥199 missense mutations) tumours compared with the 80% reference. Bars indicate 95% confidence intervals. **c**, Sensitivity for the detection of TMB-high tumours in specific cancer types (data provided are for entities with at least 10 TMB-high tumours). BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; COAD, colorectal adenocarcinoma; GBM, glioblastoma; HNSC, head and neck squamous cell carcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PANCAN, pan-cancer atlas, including all 33 cancer types analysed in TCGA; SKCM, skin cutaneous melanoma; UCEC, uterine corpus endometrial carcinoma.Bioinformatic variant filtering, which is a necessary step in the absence of paired nonmalignant DNA, is another source of technical variability. Common SNPs can be filtered out easily based on SNP databases, such as gnomAD51. However, filtering of rare or private SNPs is more error prone owing to a reliance on approximative algorithms using the detected VAF as the input. Furthermore, patient ancestry has been demonstrated to influence the accuracy of bioinformatic germline filtering methods, resulting in higher TMB values for patients who are non-white38. This bias can be explained by the content of the SNP databases used for germline filtering including a higher number of datasets from patients who are white than from others. To correct for this bias, investigators recalibrated TMB measured in the tumour-only sequencing setting based on linear models relating tumour-only TMB and tumour-germline TMB stratified by patient ancestry52. The resulting ancestry-corrected TMB outperformed classical TMB in the stratification of two retrospective cohorts for benefit from ICIs. Nevertheless, TMB at a cut-off of 10 mut/Mb remained predictive after stratification and reached statistical significance in cohorts of African, European and Asian or other backgrounds in a large retrospective cohort study53. These studies support the notion that TMB at the 10 mut/Mb cut-off is predictive of benefit from ICIs across ancestries. Nonetheless, the methods and databases currently used for germline filtering as well as the TMB cut-offs applied to specific patient populations might not be optimal and this issue warrants further investigation.
When applied to panel sequencing data, TMB needs to be approximated from the analysed part of the CDS, meaning that it is an estimate from a sample and not an exact evaluation. A stochastic error is therefore introduced that is proportional to both the inverse square root of the panel size and the inverse square root of the TMB reading provided41. For panels currently used in clinical studies and in clinical practice, the stochastic error connected with the panel size can be substantial54. The extent to which the different confounding factors contribute to the variability of TMB measurements was analysed using error modelling incorporating data on biological error, technical error, false-positive and false-negative detections of germline variants as well as stochastic error55. Panel size was the most influential source of variability for tumours with a TMB of 0–20 mut/Mb analysed using large panels, accounting for more than half of the total TMB variance. Simulating panel sequencing by subsampling WES data from patients receiving ICIs, TMB values measured using small panels (covering <0.5 Mb of the CDS) were less predictive than TMB measured using the full WES dataset, although even TMB values measured using large panels (1–1.5 Mb of the CDS) had numerically less predictive value than TMB measured using WES41. Therefore, panels including at least 1 Mb of the CDS are highly recommended for TMB measurement and the effect of using even larger panels warrants further clinical investigation.
Limitations of clinical use
Multiple studies have demonstrated a positive correlation between TMB and response to ICIs, as well as improved survival outcomes in patients with TMB-high tumours receiving ICIs compared with chemotherapy5,6,13,14,15,16,17,18,19,20,56. Nonetheless, many of these studies provide data on average outcomes across broad TMB strata and/or analysed data from multiple studies encompassing patients with several different tumour types. Other studies focusing on associations of TMB with response to ICIs in patients with specific tumour types often failed to confirm the positive associations seen in more-diverse cohorts57,58,59,60, indicating possible limitations of the clinical utility of TMB.
TMB is a continuous variable resulting in inherent challenges with categorization for clinical decision making. In particular, whether the 10 mut/Mb cut-off is optimal and equally valid across all cancer types, and the clinical consequences for a tumour having a TMB close versus far away from this cut-off, warrants further investigation. By comparing TMB-high (such as those in the top 20% quantile) versus TMB-low tumours, as well as applying TMB as a continuous variable, investigators reported significantly improved OS for patients with most cancer types with a high or higher TMB receiving ICIs, whereas TMB did not appear to be a generally prognostic feature among patients who did not receive ICIs17. These multivariate survival analyses established significant positive associations between TMB and OS for patients with multiple cancer types including NSCLC, melanoma, head and neck, colorectal, oesophagogastric and urothelial carcinomas, albeit with some exceptions such as glioma. Statistically significant associations between TMB as a continuous variable and the top 20% for each histology and improved OS persisted even when data from patients with melanoma or NSCLC were excluded from the analysis. This study supports the view that applying higher and cancer-type-specific TMB cut-offs might outperform a single fixed threshold for the stratification of patients for benefit from ICIs. In Keynote-158, which supported the entity-agnostic approval of pembrolizumab, response rates of 6.7%, 12.5% and 37% were reported for tumours with TMBs of <10, 10–13 and >13 mut/Mb, respectively, demonstrating imperfect segregation of responders at a cut-off of 10 mut/Mb (ref. 27). This cut-off was prespecified based on earlier retrospective research involving patients with NSCLC14,61. Further validation of the optimal, potentially cancer type-specific TMB cut-offs is warranted to enable more accurate selection of the patients most likely to derive benefit from ICIs.
In the previously discussed analysis comparing different TMB cut-offs, the TMB cut-off for the top 20% of patients with colorectal cancer (CRC) was particularly high (52 mut/Mb), probably reflecting the prevalence of MSI-H in this subgroup17. This study emphasized the predictive value of MMRd and POLE/POLD1 mutational status for benefit from ICIs, alongside hypermutated tumours associated with exposure to environmental carcinogens (such as UV radiation and tobacco). Nonetheless, studies focusing on MSI-H tumours found primary resistance to ICIs in some patients, indicating that MSI-H is an imperfect biomarker in this setting59. When MSI-H tumours were removed from this analysis, TMB at a cut-off of 10 mut/Mb was predictive of benefit only in a few cancer types, including metastatic head and neck cancer, NSCLC and melanoma, but not in several other malignancies. By contrast, several other studies involving patients with MSI-H cancers revealed measurable positive associations between TMB, or indel mutation load, and ICI response62,63,64,65. In a single-centre post hoc analysis, authors suggested that the subset of MSI-H and MMRd CRCs that fail to respond to ICIs might reflect misdiagnosis of MSI-H or MMR status66. The TMB of MSI-H/MMRd tumours is considerably higher than that of tumours with proficient DNA repair; therefore, assessments of TMB could be used as a quality control method and support the avoidance of misdiagnoses in this context. In summary, the current tumour-agnostic FDA Accelerated Approval of pembrolizumab at the TMB cut-off of ≥10 mut/Mb might be overly broad, suggesting a need to consider not only the absolute TMB but also the underlying cancer type.
The predictive value of TMB can depend on the specific treatment regimen under consideration and was observed to be retained with the addition of CTLA4 blockade to blockade of the PD-1–PD-L1 axis11,13,14. By contrast, for patients receiving combinations of ICIs and chemotherapy, TMB can completely lose its predictive value as observed in patients with NSCLC enrolled in Keynote-189, Keynote-407 and Keynote-21 (refs. 67,[68](#ref-CR68 "Langer, C. et al. OA04.05 KEYNOTE-021: TMB and outcomes for carboplatin and pemetrexed with or without pembrolizumab for nonsquamous NSCLC. J. Thorac. Oncol. https://doi.org/10.1016/j.jtho.2019.08.426
(2019)."),[69](/articles/s41571-024-00932-9#ref-CR69 "Garassino, M. C. et al. Evaluation of blood TMB (bTMB) in KEYNOTE-189: pembrolizumab (pembro) plus chemotherapy (chemo) with pemetrexed and platinum versus placebo plus chemo as first-line therapy for metastatic nonsquamous NSCLC. J. Clin. Oncol. 38, 9521–9521 (2020).")). This problem is not specific to TMB, but also affects several other immune-related biomarkers, including tissue-based (such as PD-L1) and blood-based biomarkers (such as lymphocyte-to-neutrophil ratio and the advanced lung cancer inflammation index), all of which lose their predictive power when chemotherapy is administered alongside an ICI[70](/articles/s41571-024-00932-9#ref-CR70 "Mountzios, G. et al. Association of the advanced lung cancer inflammation index (ALI) with immune checkpoint inhibitor efficacy in patients with advanced non-small-cell lung cancer. ESMO Open 6, 100254 (2021)."). In general, when both therapies are combined, discerning whether response and resistance are related to chemotherapy, ICIs or both becomes considerably more difficult and makes biomarker analyses more challenging.In summary, TMB has both limited sensitivity and specificity for the prediction of benefit from ICIs. To mitigate these limitations, several strategies were investigated: replacing TMB with tumour neoantigen burden (TNB) or HLA-corrected TMB to address the varying immunogenicity of the underlying mutations; refining the classical TMB to incorporate certain novel concepts including cTMB, persistent TMB (pTMB) and mutational signatures; and combining TMB with other biomarkers, such as PD-L1, MSI-H, STK11/KEAP1 mutations, derived lymphocyte-to-neutrophil ratio and/or immune-related gene expression signatures to improve the predictive validity of TMB in patients receiving chemoimmunotherapy regimens71 (Fig. 4).
Fig. 4: Concepts for the refinement of tumour mutational burden.
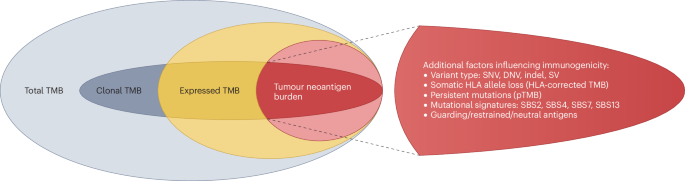
The immunological relevance of genetic alterations is governed by their expression, the processing of the mutated proteins to neopeptides and subsequent presentation as well as the recognition features of the processed neopeptides. The subgroups depicted here consider these properties for a refined version of the total tumour mutational burden (TMB). Clonal TMB focuses on truncal mutations representing alterations that are shared by all cancer cells. Hence, clonal neoantigens can be used to recognize all cells originating from a specific tumour. Expressed TMB captures the mutational burden of expressed genes and includes only the alterations manifest at the mRNA level. Tumour neoantigen burden comprises neoepitopes arising from expressed somatic mutations that result in altered peptide sequences and are predicted to be recognized by the immune system. The coloured areas correspond to expressed (yellow), neoantigenic (red) and remaining mutations (grey). Full and pale tint refers to clonal and subclonal mutations, respectively. DNV, dinucleotide variant; pTMB, persistent TMB; SBS, single-base substitution; SNV, single nucleotide variant; SV, structural variant.
Tumour neoantigen burden
TMB is used as a surrogate for the presence of neoantigens, although not all mutation types are equivalent in their capacity to produce immunogenic neoantigens. As an example, unlike missense mutations, which only affect a single amino acid, frameshift mutations completely change the protein sequence up to the next stop codon and are therefore likely to result in a higher number of neoepitopes43,72. Frameshift mutations can result in the production of virtually random peptide sequences with a high degree of dissimilarity to germline epitopes that might offer the opportunity for specific antitumour immune response, although whether the mRNAs resulting from these alterations are vulnerable to nonsense-mediated decay or other post-transcriptional processes needs to be considered. TNB involves refining TMB by specifically quantifying the subset of mutations that result in immunogenic neopeptides or the sum total of immunogenic neopeptides73.
Somatic mutations trigger the specific immune recognition of the cancer cells that present their related neoantigens, although antigen presentation is dependent on fully functioning antigen processing and presentation pathways, as well as sufficient T cell priming, expansion and antigen recognition (Fig. 1b–d). Non-synonymous mutations in the CDS of the tumour genome result in the expression of mutated proteins that can act as neoantigens. Mutated proteins must first be processed by the proteasome, giving rise to neopeptides that can be presented by MHC class I or class II molecules and promote a tumour-specific immune response74,75. MHC class I molecules are expressed by all nucleated cells and present peptides of 8–12 amino acids in length to CD8+ T cells76. MHC class II molecules are expressed predominantly by professional APCs, such as dendritic cells, and present peptides of 13–25 amino acids in length to CD4+ T cells77,78. To elicit an antitumour immune response, neoantigens need to be: expressed, processed and transported to the ER; presented by MHC molecules and recognized by T cells. To address these three steps, in silico methods have been developed for predicting antigen processing, antigen-binding affinity and antigen recognition. Bioinformatic predictors are available and can successfully address the first two of these steps using machine learning with binding affinity data and/or mass spectrometry-identified HLA-eluted ligands as training data79,80. Large datasets on antigen processing or on the combination of antigen processing and binding affinity are publicly available, for example, from the Immune Epitope Database (IEDB)81, the SysteMHC Atlas82 and the Proteomics Identification Database (PRIDE)83, whereas data on antigen recognition are scarce owing to a lack of effective high-throughput methods of analysis.
To illustrate the state of the art for computational neoantigen prediction, we analysed mutations present in tumours included in TCGA pan-cancer cohort using four published neoantigen prediction tools, all of which are based on artificial neuronal networks, to predict the binding of peptides to MHC class I molecules (Fig. 5). NetMHC 4.0 and MHCFlurry 1.2 are allele-specific predictors trained separately for each HLA type84,85. These tools predict antigen-binding affinity as a half-maximal inhibitory concentration (IC50) and separate binding antigens from non-binding antigens based on a threshold of 500 nM. By contrast, MHCPan 4.1 and MHCFlurry 2.0 are pan-cancer predictors trained with both HLA and peptide sequence as the input86,87. For these tools, two cut-offs were analysed corresponding to weak binders (cut-off 0.5%) and strong binders (cut-off 2%), respectively. For all of the neoantigen prediction approaches analysed, the correlation between TNB and TMB in the pan-cancer TCGA cohort was moderate to strong (Spearman correlation 0.65–0.79).
Fig. 5: Immunogenicity of mutations and tumour neoantigen burden.
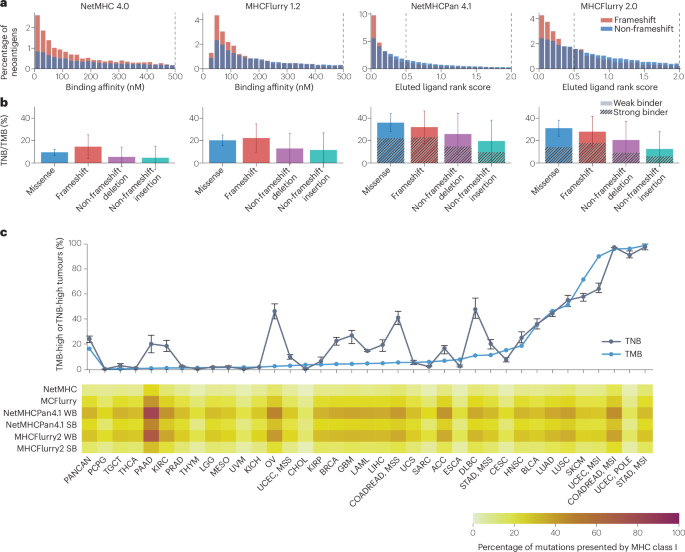
The mutations present in data from TCGA pan-cancer cohort were analysed and assessed for presentation by MHC class I using four neoantigen prediction algorithms. a, Distribution of binding affinity or eluted ligand rank scores separated by frameshift and non-frameshift mutations. Vertical lines indicate the cut-offs for binding to HLA class I molecules. For NetMHC 4.0 and MHCFlurry 1.2, the cut-off is at IC50 = 500 nM. For NetMHCPan 4.1 and MHCFlurry 2.0, two cut-offs corresponding to weak binders (WBs, 0.5%) and strong binders (SBs, 2%) were analysed. b, Percentage of mutations presented by MHC class I separately for missense mutations, frameshift mutations, non-frameshift deletions and non-frameshift insertions. Data presented are means and standard deviations across all analysed tumours. Tumour neoantigen burden (TNB) and tumour mutational burden (TMB) are moderately to strongly correlated (Spearman correlation in the pan-cancer cohort: NetMHC 4.0: 0.67, MHCFlurry 1.2: 0.65, NetMHCPan 4.1 WB: 0.79, NetMHCPan 4.1 SB: 0.68, MHCFlurry 2.0 WB: 0.79 and MHCFlurry 2.0 SB: 0.79). c, Top, Percentage of TMB-high (≥199 missense mutations) and TNB-high (TMB cut-off transferred to TNB by maximizing the Youden index) tumours per cancer type. Error bars (minimum to maximum percentage) refer to the results of six neoantigen prediction methods. Bottom, Percentage of presented mutations relative to all mutations (TNB/TMB). Data presented are mean values for each specific tumour type. ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colorectal adenocarcinoma; COADREAD, colorectal adenocarcinoma; DLBC, diffuse large B cell lymphoma; ESCA, oesophageal carcinoma; GBM, glioblastoma; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukaemia; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PANCAN, pan-cancer atlas, including all 33 cancer types analysed in TCGA; PCPG, pheochromocytoma and paraganglioma; POLE, polymerase ε; PRAD, prostate adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumour; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma. MSI, microsatellite instable; MSS, microsatellite stable.
To simulate a TNB-based classification system and to compare it with the established TMB-based classification system, the TMB cut-off of 199 missense mutations (corresponding to 10 mut/Mb) can be transferred to a TNB cut-off by maximizing the Youden index (the sum of sensitivity and specificity) in the pan-cancer cohort. For all investigated algorithms, the percentage of mutations predicted to be presented by MHC I per tumour varied significantly between cancer types (all P < 1 × 10−100). As a consequence, the percentage of tumours classified as TMB-high and as TNB-high differed markedly for certain cancer types (Fig. 5c). Thus, applying a pan-cancer cut-off for TNB instead of TMB results in substantial differences in tumour classification and, as a consequence, in the selection of patients to receive ICIs.
HLA-corrected TMB involves correcting TMB according to potential loss of HLA alleles by filtering the mutations for recognizability by the HLA alleles available in cancer cells88. HLA-corrected TMB outperformed classical TMB for the prediction of PFS in patients with NSCLC or melanoma in receiving ICIs in independent cohorts, although further data are needed to confirm the clinical utility of HLA-corrected TMB in these cancers and beyond. The differential agretopicity index89, MUC16 neoantigens90 and the neoantigen-fitness model91 have all been suggested as predictive markers for ICI response, although none were confirmed in a subsequent meta-analysis including more than 1,000 patients with melanoma as well as those with head and neck, urothelial, renal, lung and colorectal cancers92. An extended version of the neoantigen-fitness model, modelling recognition (non-self-antigens) and tolerance (self-antigens) as two competing features, was able to predict immunoediting in long-term survivors of pancreatic cancer93. In a study conducted by the TESLA consortium, a metric including neoantigen presentation and recognition features outperformed metrics exclusively based on neoantigen presentation in a cohort of 55 patients with melanoma receiving anti-PD-1 antibodies94. Although this study was one of the first to define a predictive biomarker including neoantigen recognition features, the conclusions are limited by the investigation of only 608 neoepitopes from only 6 tumours. Phase I and II trials have provided the first clinical proof of principle supporting the use of vaccines comprising mRNAs encoding personalized neoantigens in patients with pancreatic cancer or melanoma95,96. Both studies rely on computational prediction of the most potent neoantigens and demonstrate that these approaches are feasible and potentially effective, indicating an urgent need for further development.
So far, much of the research on targets for ICIs has focused on neoantigens arising from tumour-specific somatic mutations detected using DNA sequencing. Nonetheless, other large classes of tumour-specific and tumour-associated antigens exist and can potentially be profiled using RNA sequencing and/or other ‘omics’ technologies that warrant deeper investigation. These antigens arise from genomic ‘dark matter’ describing alterations that cannot be detected by WES together with the bioinformatic workflow of somatic variant calling and include chimeric proteins arising from gene fusions, novel splice isoforms, RNA-edited transcripts and transposable elements (reviewed in detail elsewhere97). Over the past few years, endogenous retrovirus expression has been suggested as a predictive biomarker and immunotherapy target in patients with several cancer types, including renal clear cell carcinoma and NSCLC98,99. The hunt for immunogenic dark matter is an exciting area of research that might enable the development of novel immunotherapies in combination with corresponding biomarkers.
Clinical evidence supporting the superiority of TNB and related biomarkers over TMB as a predictor of response to ICIs is currently limited. The still ongoing development of computational models capable of accurately predicting potent neoantigens, which in turn is limited by the amount of available training data, is an important factor contributing to this limitation. Data on the recognition of specific neoantigens by T cells are particularly scarce for these reasons. More and larger datasets will need to be produced and shared to improve neoantigen prediction capabilities and thus develop TNB into a more accurate predictive marker capable of guiding treatment decision-making in clinical practice.
TMB modifications
Several TMB-related biomarkers, including certain subsets of mutations, have been introduced with the aim of optimizing the prediction of benefit from ICIs (Box 2). The total TMB of a tissue sample can be decomposed into a sum of clonal and subclonal mutations, the former constituting the cTMB. In the previously mentioned large meta-analysis including more than 1,000 patients who received ICIs, both higher cTMB and higher total TMB were associated with an improved response to ICIs, whereas subclonal TMB was not predictive92. Nonetheless, these results should be interpreted in the context of potential limitations, including that this analysis was based on aggregated raw sequencing and RECIST data from 12 published studies. This approach might introduce artefacts from batch effects and is limited by differing levels of data availability for different cancer types as well as a lack of assessment of long-term end points. Two studies involving patients with NSCLC and melanoma support the hypothesis that ICI response is driven predominantly by cTMB, whereas higher levels of subclonal genetic diversity can be detrimental100,101. The predictive value of cTMB was investigated further in studies involving patients with advanced-stage urothelial carcinoma, MMRd gastric and colorectal cancers or NSCLC102,103,104. Data from patients with MMRd gastric or colorectal cancers provide some evidence that cTMB outperforms classical TMB. By contrast, other data do not support the use of cTMB: in patients with melanoma, cTMB did not outperform total TMB in the prediction of OS105. In a retrospective analysis of data from more than 3,000 patients with advanced-stage NSCLC receiving ICIs, subclonal TMB, but not cTMB and total TMB, separated long-term responders from short-term responders20.
We and others have previously shown that subclonal TMB estimated either bioinformatically100,101 or by sequencing two or more samples from the same tumour49 varies significantly in different regions of the tumour and consequently influences the estimation of total TMB in a sample-dependent manner. Thus, applying cTMB instead of total TMB might ameliorate the effects of this biological variability. Sequencing multiple samples from the same tumour and/or single-cell sequencing can provide deeper insights into the clonality of mutations, although cTMB can also be approximated bioinformatically based on sequencing data from a single tumour sample92,102. Compared with classical TMB, cTMB includes only truncal mutations, which typically have higher VAFs and can therefore be detected at a higher level of sensitivity, potentially providing a more-robust biomarker. Although the argument of providing a potentially more-robust biomarker as well as some of the studies analysing clinical correlates favour cTMB, the overall level of evidence is currently not sufficient to support the clinical application of cTMB over TMB.
As another TMB modification, pTMB includes only mutations that are unlikely to be lost during tumour evolution or as a mechanism of acquired resistance106. Thus, only mutations located in genes with a single copy present in tumour DNA or harbouring multiple copies of the mutated allele are considered. pTMB has been shown to outperform TMB in the prediction of response to ICIs in patients with melanoma, head and neck cancer, mesothelioma or NSCLC106. Furthermore, persistent mutations were encountered across the entire spectrum of clonal heterogeneity, suggesting that the predictive value of pTMB is independent of that of cTMB. To the best of our knowledge, this is the only study investigating pTMB with results currently available; therefore, further studies investigating this interesting new concept that might confirm the predictive superiority of pTMB over classical TMB are warranted.
Expression of the mutated proteins is a prerequisite for presentation of the corresponding neoantigens and their recognition by the immune system. On the basis of this necessity, investigations of whether expressed TMB (eTMB) provides more accurate predictions of a response to ICIs than classical TMB are warranted. In a combined WES and whole-transcriptome sequencing analysis, eTMB outperformed TMB in the prediction of OS in patients with melanoma receiving ICIs107. Other studies revealed robust correlations between these two biomarkers, albeit without an improvement in predictive accuracy with eTMB relative to TMB108,109,110,111. Thus, although gene expression analysis is mandatory when aiming to identify potent neoantigens, for example, for personalized vaccination, further studies are needed before use of eTMB, and the additional analyses required for quantification, instead of TMB, can be justified for the prediction of response to ICIs.
Mutational signatures, which were originally introduced in 2012, involve decomposing TMB according to the underlying mutational processes that are active during tumorigenesis and tumour growth112,113,[114](#ref-CR114 "Degasperi, A. et al. Substitution mutational signatures in whole-genome-sequenced cancers in the UK population. Science https://doi.org/10.1126/science.abl9283
(2022)."),[115](/articles/s41571-024-00932-9#ref-CR115 "Nik-Zainal, S. et al. Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 (2012)."). In a review published in 2020[116](/articles/s41571-024-00932-9#ref-CR116 "Sha, D. et al. Tumor mutational burden as a predictive biomarker in solid tumors. Cancer Discov. 10, 1808–1825 (2020)."), we quantified the extent to which specific signatures contribute to TMB. This pan-cancer analysis revealed the main mutational processes associated with hypermutation and a high TMB: _POLE_/_POLD1_ mutations, MMRd, UV light exposure, tobacco smoking, activation of activation-induced deaminase/APOBEC and those associated with the two clock-like mutational processes (single-base substitution (SBS)1 and SBS5). The current version of the COSMIC Mutational Signatures (3.4) includes a total of 67 SBS and 23 indel signatures[117](/articles/s41571-024-00932-9#ref-CR117 "The COSMIC Database v3.4.
https://cancer.sanger.ac.uk/signatures/
(2023)."). From the clinical perspective, discerning whether the mutational signatures associated with targetable biological alterations can be exploited as predictive biomarkers will be an important step.Mutational signatures have been suggested as biomarkers of various specific DNA repair deficiencies, including MMRd (SBS6, SBS14, SBS15, SBS20, SBS21, SBS26 and SBS44), homologous recombination repair deficiency (HRD) (SBS3), PRD (SBS10a–d, SBS14 and SBS20) and base-excision repair deficiency (SBS36)[117](/articles/s41571-024-00932-9#ref-CR117 "The COSMIC Database v3.4. https://cancer.sanger.ac.uk/signatures/
(2023)."). HRD-associated mutational signatures have been used to identify patients, mostly with breast or ovarian cancers, who are most likely to benefit from DNA-damage-inducing agents or PARP inhibitors[118](/articles/s41571-024-00932-9#ref-CR118 "Zhao, E. Y. et al. Homologous recombination deficiency and platinum-based therapy outcomes in advanced breast cancer. Clin. Cancer Res. 23, 7521–7530 (2017)."). A high level of accuracy in separating patients with HRD-deficient cancers from those with HRD-proficient cancers is achieved using complex whole-genome sequencing (WGS)-based classifiers including larger sets of features such as SBSs, indels, genomic rearrangements and CNAs (such as CHORD and HRDetect)[119](/articles/s41571-024-00932-9#ref-CR119 "Davies, H. et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 23, 517–525 (2017)."),[120](/articles/s41571-024-00932-9#ref-CR120 "Nguyen, L., Martens, J. W. M., Van Hoeck, A. & Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 11, 5584 (2020)."). In an analysis of WGS data from 129 patients with oesophageal cancer, investigators subdivided patients based on SBSs and mutational signatures and hypothesized that those with HRD-positive cancers might benefit from PARP inhibitors[121](/articles/s41571-024-00932-9#ref-CR121 "Secrier, M. et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet. 48, 1131–1141 (2016)."). Indeed, HRD mutational signatures are able to discriminate between xenografts that respond and that do not respond to PARP inhibitors in patients with gastric cancer[122](/articles/s41571-024-00932-9#ref-CR122 "Petrelli, A. et al. BRCA2 germline mutations identify gastric cancers responsive to PARP inhibitors. Cancer Res. 83, 1699–1710 (2023)."). However, tumour evolution during and in response to therapy needs to be considered when using genomic biomarkers as the functionality of deficient DNA repair can be reinstated, exemplified by the difference between ongoing and historic HRD[123](/articles/s41571-024-00932-9#ref-CR123 "Peng, G. et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat. Commun. 5, 3361 (2014)."). Restoration of previously deficient DNA repair mechanisms can occur as a response to the selection pressures exerted by therapeutic interventions, such as reversal mutations in _BRCA1/2_ observed in relevant proportions of patients with breast cancer or patients with prostate cancer with acquired resistance to PARP inhibitors[124](/articles/s41571-024-00932-9#ref-CR124 "Li, H. et al. PARP inhibitor resistance: the underlying mechanisms and clinical implications. Mol. Cancer 19, 107 (2020).").Second, specific mutational signatures have been proposed as biomarkers to predict benefit from immunotherapies. The most prominent example is provided by mutational signatures associated with dMMR, which is a robust and approved biomarker of response to ICIs across solid tumours125,126,127. Factors mediating the responses of these tumours to ICIs include the high missense mutational burden and especially the high indel burden owing to the highly immunogenic features of frameshift neoantigens43. Of note, in an analysis of sequencing data from more than 1,500 gliomas, dMMR was not predictive of ICI response possibly owing to an absence of potent clonal neoantigens, whereas many of the mutations generated by dMMR were subclonal128. The mutational signatures for tobacco smoking (SBS4) as well as APOBEC activation were both associated with a higher neoantigen burden and subsequently with increased sensitivity to anti-PD-1 antibodies in patients with NSCLC5,129,130,131. The positive correlation between APOBEC mutational signature and efficacy of ICIs is supported by further studies with mixed cohorts of patients with solid tumours130,132. Stratification of patients with melanoma according to the dominant mutational signature revealed that, in comparison to other signatures, the UV exposure and alkylating agent subgroups had higher ORRs (including higher proportions of both complete and partial responses) to ICIs130. In another study, ICIs were found to be less effective in patients with NSCLC and melanoma with high activity of the clock-like signature SBS1133. In contrast to NSCLC, the tobacco smoking mutational signature was associated with an inferior response to ICIs in patients with head and neck squamous cell carcinoma134. These various data demonstrate that mutational signatures alone cannot predict ICI outcome in a tumour-agnostic manner. In summary, considering tumour entity and the underlying mutational processes in addition to classical TMB can provide more accurate guidance on use of ICIs.
A major barrier to the introduction of mutational signatures as diagnostic tools in routine clinical practice is the broad sequencing approach needed for sensitive detection. This requirement is a consequence of the classification of the mutations into 96 features (6 types of mutation plus the bases located immediately before and after the mutation) that need to be identified and counted separately4. Low feature counts will increase the corresponding stochastic errors. Thus, WGS is the optimal approach for the determination of mutational signatures, whereas WES is less sensitive and typical panel-based sequencing approaches will only be feasible for the detection of highly abundant signatures135. However, WGS comes with substantially higher labour requirements and costs as well as greater data storage and processing demands. In the coming years, these limitations will probably be mitigated by the continuing trend of falling sequencing costs and microchip prices[136](/articles/s41571-024-00932-9#ref-CR136 "NIH. The cost of sequencing a human genome. genome.gov https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost
(2021).") and will probably create opportunities for broader applications of WGS and mutational signatures in oncology practice.Combinations with other biomarkers
In melanoma, TMB has been combined with transcriptomic and immune cell omics data, or with inflammatory gene expression signatures and BRAF mutational status to create multimodal predictors, and both approaches have been shown to be superior to TMB alone in identifying patients who are likely to benefit from ICIs107,137. Data from one study also indicate that the predictive validity of TMB is dependent on melanoma subtype and, in addition, that multiple genomic and transcriptomic parameters, including neoantigen presentation by MHC I and II complexes, are predictive of outcome on ICIs138. Similar approaches have been applied in other cancer types including NSCLC19,50, urothelial carcinoma102 or pleural mesothelioma139. Thus, the combination of TMB, gene-expression profiling, analysis of the tumour microenvironment, immune cell composition and marker expression is considered to be superior to the estimation of TMB alone92,140,141,142,143,144,145,146,147. This view is in line with the conclusions of a community challenge designed to leverage crowdsourcing to generate models capable of predicting clinical outcomes on ICIs in patients with NSCLC. This initiative, which included 417 models from 59 teams, demonstrated that models including TMB and PD-L1 plus optional additional gene expression signatures outperformed predictions based on TMB and PD-L1 only148.
In another approach, somatic mutations in specific genes or pathways are combined with TMB. In this regard, the CIRCLE algorithm outperformed TMB alone for the prediction of response to ICIs in patients with various solid tumours149. CIRCLE is a logistic model combining TMB, mutations in the genes BCLAF1, KRAS, BRAF and TP53 (which affect pathways such as MAPK and immunomodulatory signalling), age and cancer type. Up until 2023, a lack of evidence existed on the implications of a high TMB (≥10 mut/Mb) for patients with microsatellite-stable (MSS) gastrointestinal tumours. In an attempt to address this lack of evidence, investigators developed a modified TMB model including the mutation status of 16 genes to predict responsiveness to ICIs in patients with these tumours (comprising 3.29% of a large, retrospective dataset)150. Patients with MSS gastrointestinal cancers with mutations in any gene included in the modified TMB signature had superior OS in response to ICIs. These studies support the view that combining TMB with mutations in driver genes or those involved in other altered oncogenic signalling pathways might improve predictive validity.
Additional parameters have been suggested for the prediction of responsiveness to ICIs in patients with MSI-H cancers. A study published in 2023 reported significantly improved PFS, but not OS, in patients with metastatic MSI-H CRCs of hereditary (such as from Lynch syndrome) compared with sporadic origins151, potentially reflecting a higher TMB in the former compared with the latter group152. Although neither _BRAF_V600E nor RAF mutations predicted PFS or OS in patients with metastatic MSI-H CRC receiving ICIs151, more-complex DNA-based and RNA-based signatures have been proposed as predictors of benefit in these patients153. Finally, data published in 2023 indicate that clonal rather than subclonal TMB is predictive of ICI response in patients with MSI-H gastric cancers and CRCs, indicating a role of intratumoural heterogeneity103.
Data from most studies demonstrate a positive correlation among TMB, TNB and immune-cell infiltration. Nonetheless, presentation of mutation-induced antigens as evaluated by TMB or TNB requires functioning mechanisms of antigen presentation, T cell expansion and antigen recognition, as well as a tumour microenvironment that permits immune–tumour cell interactions. In patients with _IDH_-wild-type glioma, TNB alone did not correlate with outcome but became predictive when combined with the infiltration of CD8+ T cells154. Scenarios in which TMB-low tumours expressing a small number of highly immunogenic antigens elicit a robust cytotoxic T cell response have also been documented57.
Most importantly, alterations in the antigen processing and presentation machinery need to be accounted for in studies examining ICI response as such alterations can alter local immune responses and confer resistance to ICIs. Resistance to ICIs in patients with melanoma has previously been linked with defects in the antigen presentation machinery, often arising from loss-of-function mutations in B2M155. B2M has a crucial role in the assembly of the MHC class I antigen complex and cancer cells harbouring such mutations cannot effectively present antigens to cytotoxic T cells156. MSI-H CRCs had the highest frequency of inactivating B2M mutations seen among any cancers157, probably reflecting the massive immune-mediated selective pressures occurring during the development of these tumours43. Interestingly, _B2M_-mutant MSI-H cancers have been reported to have similar levels of sensitivity to ICIs relative to their _B2M_-wild-type counterparts[158](/articles/s41571-024-00932-9#ref-CR158 "Middha, S. et al. Majority of B2M-mutant and -deficient colorectal carcinomas achieve clinical benefit from immune checkpoint inhibitor therapy and are microsatellite instability-high. JCO Precis. Oncol. https://doi.org/10.1200/PO.18.00321
(2019)."). This surprising observation might be explained by the small cohort size and retrospective design of this study, with other studies suggesting that such mutations might even confer a more-favourable prognosis[159](#ref-CR159 "Tikidzhieva, A. et al. Microsatellite instability and beta2-microglobulin mutations as prognostic markers in colon cancer: results of the FOGT-4 trial. Br. J. Cancer 106, 12391245 (2012)."),[160](#ref-CR160 "Barrow, P. et al. Confirmation that somatic mutations of beta-2 microglobulin correlate with a lack of recurrence in a subset of stage II mismatch repair deficient colorectal cancers from the QUASAR trial. Histopathology 75, 236–246 (2019)."),[161](/articles/s41571-024-00932-9#ref-CR161 "Busch, E. et al. Beta-2-microglobulin mutations are linked to a distinct metastatic pattern and a favorable outcome in microsatellite-unstable stage IV gastrointestinal cancers. Front. Oncol. 11, 669774 (2021)."). Alternatively, CD4+ and γδ T cells have been proposed as effector cells capable of rejecting cancer cells independent of B2M or HLA-I expression, respectively[162](/articles/s41571-024-00932-9#ref-CR162 "Germano, G. et al. CD4 T cell-dependent rejection of beta-2 microglobulin null mismatch repair-deficient tumors. Cancer Discov. 11, 1844–1859 (2021)."),[163](/articles/s41571-024-00932-9#ref-CR163 "de Vries, N. L. et al. γδ T cells are effectors of immunotherapy in cancers with HLA class I defects. Nature 613, 743–750 (2023)."). Besides defects in the antigen-processing machinery, impaired IFNγ signalling, for example, owing to loss-of-function mutations in _JAK1_ or _JAK2_ can affect responsiveness to ICIs[164](/articles/s41571-024-00932-9#ref-CR164 "Marabelle, A., Aspeslagh, S., Postel-Vinay, S. & Soria, J. C. JAK mutations as escape mechanisms to anti-PD-1 therapy. Cancer Discov. 7, 128–130 (2017)."). Alterations in these pathways are not only of major importance for interpreting the effects of TNB but also have profound implications for the immunobiology of the affected cells and should be considered when attempting to understand the relationship between TNB and ICI response.Future perspectives
Accumulating evidence supports the clinical application of TMB as a predictive biomarker of responsiveness to ICIs in patients with solid tumours. Higher levels of clinical benefit from ICIs can be observed across several different TMB-high cancer types, although evidence supporting the predictive value of TMB in specific cancer types is weaker and only applies for a subset of entities. In a retrospective analysis including data from almost 2,000 patients who received ICIs, cancer types were grouped into entities showing significant positive correlations between CD8+ T cell infiltrates and TNB (category I including endometrial cancer, melanoma, lung adenocarcinoma, cervical cancer, bladder cancer and colorectal cancer) and those that did not (category II)60. In category II cancer types, a TMB cut-off of 10 mut/Mb failed to predict a response to ICIs, supporting the view that further evaluations of TMB in specific cancer types as well as optimization of cut-offs is warranted.
In 2020, on the basis of data from Keynote-158, pembrolizumab was approved by the FDA for patients with TMB-high tumours (≥10 mut/Mb), regardless of the tissue of origin following disease progression on one or more previous treatments and who have no satisfactory alternative treatment options available. So far, no corresponding approval for use in Europe has been reported. Clinical trial data from the past few years support the utility of TMB in CUP, a diagnosis that comprises a heterogeneous group of diseases potentially including several different cancer types often with limited treatment options28,29. Outside regulatory approvals, such as in Europe, TMB can warrant discussion in the setting of late-line off-label treatments, when all available approved therapies have been exhausted. To further expand clinical utility, TMB should be further investigated in retrospective and prospective studies defining a single or disease-specific cut-off and in combination with other biomarkers.
The main confounders of the accuracy of TMB measurements include the use of small NGS panels and the limited purity of some tumour tissue samples. The use of smaller panels results in less-precise measurements of TMB and inferior separation of patients responding and not responding to ICIs; therefore, panel sizes of at least about 1 Mb are mandatory for clinical measurements of TMB41. Of note, even for panel sizes of 1 Mb, the level of stochastic imprecision will still substantially affect the accuracy of TMB quantification in tumours with a low-to-medium TMB (≤20 mut/Mb)55. Thus, investigations of larger sequencing panels (≥2 Mb) or WES for improved clinical performance are warranted. Analysis of samples with low tumour purity will reduce sensitivity for the detection of somatic variants and, in turn, lead to lower TMB values. As a consequence, the sensitivity to detect patients with a high TMB will be reduced. This loss of sensitivity might result in patients who are likely to benefit from ICIs not receiving these agents and, in parallel, can lead to false-negative results in clinical trials comparing patients with TMB-high tumours with those with TMB-low tumours. Tumour purity ≥60% is optimal and tumour purity in the range of 40–60% leads to a moderate reduction in sensitivity; however, such reductions become particularly substantial below 40%. In summary, obtaining samples with a high level of tumour purity is crucial for accurate TMB measurements and should be maximized using careful selection of histopathological samples as well as tumour cell enrichment strategies.
The complexity of TMB and its measurement as well as the even greater complexity of TMB modifications and extensions requires broad sequencing and the application of increasingly sophisticated bioinformatic tools (for example, for the prediction of immunogenicity of mutations), which can be a major barrier to clinical implementation. A study on the clinical implementation of biomolecular technologies across Europe in 2023 described the heterogenous availability of comprehensive sequencing approaches, such as large NGS panels, WES and WGS165. Achieving the aim of better separation of ICI responders and non-responders will require more accurate diagnostic tools owing to the inherent complexity of the underlying immunobiology. This demand for better biomarkers and the increasing efforts and expertise needed to develop and implement the related assays in clinical practice create an area of conflict that needs to be addressed. In the coming years, limitations in the availability of NGS and bioinformatic expertise will probably be mitigated by the continuation of trends such as falling sequencing costs and microchip prices as well as national clinical sequencing projects and/or the centralization of molecular diagnostics at specialized hubs[166](/articles/s41571-024-00932-9#ref-CR166 "Genomics England. The 100,000 Genomes Project. https://www.genomicsengland.co.uk/initiatives/100000-genomes-project
(2024)."),[167](/articles/s41571-024-00932-9#ref-CR167 "The German Federal Ministry of Health. GenomeDE — National Strategy for Genomic Medicine.
https://www.bundesgesundheitsministerium.de/en/en/international/european-health-policy/genomde-en.html
(2024).").Besides TMB, PD-L1 expression and MSI-H are included in the approvals of ICIs for certain groups of patients. Nonetheless, TMB and PD-L1 have been shown not to correlate in most cancer types and are often independently predictive of a response to ICIs. TMB has also been demonstrated to have predictive value both in patients with MSS and MSI-H cancers, although the added predictive value of TMB in tumours accurately identified as MSI-H is often limited. Thus, strategies aiming to optimally combine TMB with the other biomarkers need to be developed and validated. For scenarios in which targeting an actionable molecular alteration as well as administering an ICI is feasible, prioritization or even combination of therapies needs to be addressed. In patients with lung adenocarcinoma, most actionable driver alterations, including EGFR mutations and ALK, ROS or RET fusions (but not _KRAS_G12C mutations), are characterized by limited immunogenicity. Targeted therapies are the preferred first-line strategy for most of these patients, although the use of ICIs in later lines is a matter of debate. In other cancer types and with other targeted therapies, clinical data on how to proceed when both targeted therapy and ICIs are indicated are often incomplete, and further studies designed to evaluate the prioritization of one class of agent over another, and/or the feasibility of combining therapies targeting actionable mutations with ICIs guided by biomarkers such as TMB, are warranted.
Modifications of TMB either focusing on specific subsets of mutations or incorporating the immunogenicity of specific classes of mutations (such as cTMB, pTMB, eTMB, TNB and HLA-corrected TMB) have all outperformed classical TMB according to the data from published studies. However, most of these studies evaluated only one of these concepts in a single cohort, thus hindering direct comparisons of performance. Therefore, further validation will be necessary before these concepts can enter clinical practice. Such validations should include head-to-head comparisons of multiple biomarkers in large retrospective cohorts, as seen already in a large meta-analysis92, with subsequent prospective validation of the most clinically relevant observations.
Conclusions
Data from multiple studies support the predictive validity of TMB as a biomarker of benefit from ICIs within and across cancer types. These observations, together with the inclusion of TMB in the entity-agnostic approval of pembrolizumab, have led to the introduction of TMB into clinical practice. Nevertheless, two major limitations remain and need to be addressed in further research: (1) the predictive utility of TMB at the 10 mut/Mb cut-off has been demonstrated only for some and not all cancer types. (2) For all of the investigated cancer types, the ability of TMB to discriminate between patients who respond and those who do not respond to ICIs is imperfect. In this context, remembering that somatic mutations are the first constituent in a long chain of factors triggering cell-mediated antitumour immunity is important (Fig. 1). Additional factors essential for this process include antigen processing and presentation, T cell priming as well as recognition and killing of cancer cells by cytotoxic T cells. A more holistic and comprehensive evaluation of these factors including the presence of loss-of-function alterations in antigen processing and presentation pathways as well as combining alterations affecting these various processes into a signature will be key to overcoming the limited sensitivity and specificity of TMB for the prediction of a response to ICIs.
References
- Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Article CAS PubMed Google Scholar - Vogelstein, B. & Kinzler, K. W. The multistep nature of cancer. Trends Genet. 9, 138141 (1993).
Article Google Scholar - Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Article CAS PubMed PubMed Central Google Scholar - Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Article CAS PubMed PubMed Central Google Scholar - Rizvi, N. A. et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Article CAS PubMed PubMed Central Google Scholar - Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014).
Article PubMed PubMed Central Google Scholar - Gandhi, L. et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 378, 2078–2092 (2018).
Article CAS PubMed Google Scholar - Paz-Ares, L. et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med. 379, 2040–2051 (2018).
Article CAS PubMed Google Scholar - Mok, T. S. K. et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet 393, 1819–1830 (2019).
Article CAS PubMed Google Scholar - Hellmann, M. D. et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N. Engl. J. Med. 381, 2020–2031 (2019).
Article CAS PubMed Google Scholar - Chan, T. A. et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol. 30, 44–56 (2019).
Article CAS PubMed Google Scholar - Carbone, D. P. et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N. Engl. J. Med. 376, 2415–2426 (2017).
Article CAS PubMed PubMed Central Google Scholar - Hellmann, M. D. et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 33, 843–852.e4 (2018).
Article CAS PubMed PubMed Central Google Scholar - Hellmann, M. D. et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 378, 2093–2104 (2018).
Article CAS PubMed PubMed Central Google Scholar - Yarchoan, M., Hopkins, A. & Jaffee, E. M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 377, 2500–2501 (2017).
Article PubMed PubMed Central Google Scholar - Banchereau, R. et al. Molecular determinants of response to PD-L1 blockade across tumor types. Nat. Commun. 12, 3969 (2021).
Article CAS PubMed PubMed Central Google Scholar - Samstein, R. M. et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 51, 202–206 (2019).
Article CAS PubMed PubMed Central Google Scholar - Cao, D., Xu, H., Xu, X., Guo, T. & Ge, W. High tumor mutation burden predicts better efficacy of immunotherapy: a pooled analysis of 103078 cancer patients. Oncoimmunology 8, e1629258 (2019).
Article PubMed PubMed Central Google Scholar - Ricciuti, B. et al. Association of high tumor mutation burden in non-small cell lung cancers with increased immune infiltration and improved clinical outcomes of PD-L1 blockade across PD-L1 expression levels. JAMA Oncol. 8, 1160–1168 (2022).
Article PubMed PubMed Central Google Scholar - Thummalapalli, R. et al. Clinical and molecular features of long-term response to immune checkpoint inhibitors in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 20, 4408–4418 (2023).
Article Google Scholar - Yarchoan, M. et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 4, e126908 (2019).
Article PubMed PubMed Central Google Scholar - Budczies, J. et al. Integrated analysis of the immunological and genetic status in and across cancer types: impact of mutational signatures beyond tumor mutational burden. Oncoimmunology 7, e1526613 (2018).
Article PubMed PubMed Central Google Scholar - Krämer, A. et al. Cancer of unknown primary: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 34, 228–246 (2023).
Article PubMed Google Scholar - Ross, J. S. et al. Comprehensive genomic profiling of carcinoma of unknown primary origin: retrospective molecular classification considering the CUPISCO study design. Oncologist 26, e394–e402 (2021).
Article CAS PubMed Google Scholar - Bochtler, T. et al. Prognostic impact of copy number alterations and tumor mutational burden in carcinoma of unknown primary. Genes Chromosomes Cancer 61, 551–560 (2022).
Article CAS PubMed Google Scholar - Gatalica, Z., Xiu, J., Swensen, J. & Vranic, S. Comprehensive analysis of cancers of unknown primary for the biomarkers of response to immune checkpoint blockade therapy. Eur. J. Cancer 94, 179–186 (2018).
Article PubMed Google Scholar - Marabelle, A. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 21, 1353–1365 (2020).
Article CAS PubMed Google Scholar - Pouyiourou, M. et al. Nivolumab and ipilimumab in recurrent or refractory cancer of unknown primary: a phase II trial. Nat. Commun. 14, 6761 (2023).
Article CAS PubMed PubMed Central Google Scholar - Krämer, A. et al. Molecularly guided therapy versus chemotherapy after disease control in unfavourable cancer of unknown primary (CUPISCO): an open-label, randomised, phase 2 study. Lancet 404, 527–539 (2024).
Article PubMed Google Scholar - Gandara, D. R. et al. Tumor mutational burden (TMB) measurement from an FDA-approved assay and real-world overall survival (rwOS) on single-agent immune checkpoint inhibitors (ICI) in over 8,000 patients across 24 cancer types. J. Clin. Oncol. 41, 2503 (2023).
Article Google Scholar - Davis, A. A. et al. Comparison of tumor mutational burden (TMB) across tumor tissue and circulating tumor DNA (ctDNA). J. Clin. Oncol. 35, e23028 (2017).
Article Google Scholar - Gandara, D. R. et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 24, 1441–1448 (2018).
Article CAS PubMed Google Scholar - Wang, Z. et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non-small cell lung cancer with use of a next-generation sequencing cancer gene panel. JAMA Oncol. 5, 696–702 (2019).
Article PubMed PubMed Central Google Scholar - Si, H. et al. A blood-based assay for assessment of tumor mutational burden in firstline metastatic NSCLC treatment: results from the MYSTIC study. Clin. Cancer Res. 27, 1631–1640 (2021).
Article CAS PubMed Google Scholar - Peters, S. et al. Atezolizumab versus chemotherapy in advanced or metastatic NSCLC with high blood-based tumor mutational burden: primary analysis of BFAST cohort C randomized phase 3 trial. Nat. Med. 28, 1831–1839 (2022).
Article CAS PubMed PubMed Central Google Scholar - Prasad, V. & Addeo, A. The FDA approval of pembrolizumab for patients with TMB >10 mut/Mb: was it a wise decision? No. Ann. Oncol. 31, 1112–1114 (2020).
Article CAS PubMed Google Scholar - Subbiah, V., Solit, D. B., Chan, T. A. & Kurzrock, R. The FDA approval of pembrolizumab for adult and pediatric patients with tumor mutational burden (TMB) ≥ 10: a decision centered on empowering patients and their physicians. Ann. Oncol. 31, 1115–1118 (2020).
Article CAS PubMed Google Scholar - Chang, H. et al. Bioinformatic methods and bridging of assay results for reliable tumor mutational burden assessment in non-small-cell lung cancer. Mol. Diagn. Ther. 23, 507–520 (2019).
Article CAS PubMed PubMed Central Google Scholar - Vega, D. M. et al. Aligning tumor mutational burden (TMB) quantification across diagnostic platforms: phase II of the Friends of Cancer Research TMB Harmonization Project. Ann. Oncol. 32, 1626–1636 (2021).
Article CAS PubMed Google Scholar - Stenzinger, A. et al. Harmonization and standardization of panel-based tumor mutational burden measurement: real-world results and recommendations of the quality in pathology study. J. Thorac. Oncol. 15, 1177–1189 (2020).
Article CAS PubMed Google Scholar - Budczies, J. et al. Optimizing panel-based tumor mutational burden (TMB) measurement. Ann. Oncol. 30, 1496–1506 (2019).
Article CAS PubMed Google Scholar - Turajlic, S. et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 18, 1009–1021 (2017).
Article CAS PubMed Google Scholar - Kloor, M. & von Knebel Doeberitz, M. The immune biology of microsatellite-unstable cancer. Trends Cancer 2, 121–133 (2016).
Article PubMed Google Scholar - Tomasetti, C., Vogelstein, B. & Parmigiani, G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc. Natl Acad. Sci. USA 110, 1999–2004 (2013).
Article CAS PubMed PubMed Central Google Scholar - Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013).
Article CAS PubMed PubMed Central Google Scholar - Merino, D. M. et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J. Immunother. Cancer 8, e000147 (2020).
Article PubMed PubMed Central Google Scholar - Menzel, M. et al. Multicentric pilot study to standardize clinical whole exome sequencing (WES) for cancer patients. npj Precis. Oncol. 7, 106 (2023).
Article CAS PubMed PubMed Central Google Scholar - Rosenthal, R. et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485 (2019).
Article CAS PubMed PubMed Central Google Scholar - Kazdal, D. et al. Spatial and temporal heterogeneity of panel-based tumor mutational burden in pulmonary adenocarcinoma: separating biology from technical artifacts. J. Thorac. Oncol. 14, 1935–1947 (2019).
Article CAS PubMed Google Scholar - Anagnostou, V. et al. Multimodal genomic features predict outcome of immune checkpoint blockade in non-small-cell lung cancer. Nat. Cancer 1, 99–111 (2020).
Article CAS PubMed PubMed Central Google Scholar - Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Article CAS PubMed PubMed Central Google Scholar - Nassar, A. H. et al. Ancestry-driven recalibration of tumor mutational burden and disparate clinical outcomes in response to immune checkpoint inhibitors. Cancer Cell 40, 1161–1172.e5 (2022).
Article CAS PubMed PubMed Central Google Scholar - Huang, R. S. P., Graf, R. P. & Oxnard, G. R. Not all TMB assays are the same: clinical validity of robust algorithmic germline filtering. Cancer Cell 41, 819–820 (2023).
Article CAS PubMed Google Scholar - Buchhalter, I. et al. Size matters: dissecting key parameters for panel-based tumor mutational burden analysis. Int. J. Cancer 144, 848–858 (2019).
Article CAS PubMed Google Scholar - Budczies, J. et al. Quantifying potential confounders of panel-based tumor mutational burden (TMB) measurement. Lung Cancer 142, 114–119 (2020).
Article PubMed Google Scholar - Osipov, A. et al. Tumor mutational burden, toxicity, and response of immune checkpoint inhibitors targeting PD(L)1, CTLA-4, and combination: a meta-regression analysis. Clin. Cancer Res. 26, 4842–4851 (2020).
Article CAS PubMed PubMed Central Google Scholar - Sneddon, S. et al. Identification of a CD8+ T-cell response to a predicted neoantigen in malignant mesothelioma. Oncoimmunology 9, 1684713 (2020).
Article PubMed Google Scholar - Ros, J. et al. Immunotherapy for colorectal cancer with high microsatellite instability: the ongoing search for biomarkers. Cancers 15, 4245 (2023).
Article CAS PubMed PubMed Central Google Scholar - Rousseau, B. et al. The spectrum of benefit from checkpoint blockade in hypermutated tumors. N. Engl. J. Med. 384, 1168–1170 (2021).
Article PubMed PubMed Central Google Scholar - Mcgrail, D. J. et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 32, 661–672 (2021).
Article CAS PubMed Google Scholar - Ready, N. et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J. Clin. Oncol. 37, 992–1000 (2019).
Article CAS PubMed PubMed Central Google Scholar - Kwon, M. et al. Determinants of response and intrinsic resistance to PD-1 blockade in microsatellite instability-high gastric cancer. Cancer Discov. 11, 2168–2185 (2021).
Article CAS PubMed Google Scholar - Mandal, R. et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364, 485–491 (2019).
Article CAS PubMed PubMed Central Google Scholar - Schrock, A. B. et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 30, 10961103 (2019).
Article Google Scholar - Loupakis, F. et al. Prediction of benefit from checkpoint inhibitors in mismatch repair deficient metastatic colorectal cancer: role of tumor infiltrating lymphocytes. Oncologist 25, 481–487 (2020).
Article CAS PubMed PubMed Central Google Scholar - Cohen, R. et al. Association of primary resistance to immune checkpoint inhibitors in metastatic colorectal cancer with misdiagnosis of microsatellite instability or mismatch repair deficiency status. JAMA Oncol. 5, 551–555 (2019).
Article PubMed Google Scholar - Paz-Ares, L. et al. Pembrolizumab (pembro) plus platinum-based chemotherapy (chemo) for metastatic NSCLC: tissue TMB (tTMB) and outcomes in KEYNOTE-021, 189, and 407. Ann. Oncol. 30, v917–v918 (2019).
Article Google Scholar - Langer, C. et al. OA04.05 KEYNOTE-021: TMB and outcomes for carboplatin and pemetrexed with or without pembrolizumab for nonsquamous NSCLC. J. Thorac. Oncol. https://doi.org/10.1016/j.jtho.2019.08.426 (2019).
- Garassino, M. C. et al. Evaluation of blood TMB (bTMB) in KEYNOTE-189: pembrolizumab (pembro) plus chemotherapy (chemo) with pemetrexed and platinum versus placebo plus chemo as first-line therapy for metastatic nonsquamous NSCLC. J. Clin. Oncol. 38, 9521–9521 (2020).
Article Google Scholar - Mountzios, G. et al. Association of the advanced lung cancer inflammation index (ALI) with immune checkpoint inhibitor efficacy in patients with advanced non-small-cell lung cancer. ESMO Open 6, 100254 (2021).
Article CAS PubMed PubMed Central Google Scholar - Alessi, J. V. et al. Clinicopathologic and genomic factors impacting efficacy of firstline chemoimmunotherapy in advanced NSCLC. J. Thorac. Oncol. 18, 731–743 (2023).
Article CAS PubMed PubMed Central Google Scholar - Giannakis, M. et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 15, 857–865 (2016).
Article CAS PubMed PubMed Central Google Scholar - Wang, P., Chen, Y. & Wang, C. Beyond tumor mutation burden: tumor neoantigen burden as a biomarker for immunotherapy and other types of therapy. Front. Oncol. 11, 672677 (2021).
Article CAS PubMed PubMed Central Google Scholar - Rock, K. L., Reits, E. & Neefjes, J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. 37, 724–737 (2016).
Article CAS PubMed PubMed Central Google Scholar - Neefjes, J., Jongsma, M. L., Paul, P. & Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 11, 823–836 (2011).
Article CAS PubMed Google Scholar - Jongsma, M. L. M., Neefjes, J. & Spaapen, R. M. Playing hide and seek: tumor cells in control of MHC class I antigen presentation. Mol. Immunol. 136, 36–44 (2021).
Article CAS PubMed Google Scholar - Macy, A. M., Herrmann, L. M., Adams, A. C. & Hastings, K. T. Major histocompatibility complex class II in the tumor microenvironment: functions of nonprofessional antigen-presenting cells. Curr. Opin. Immunol. 83, 102330 (2023).
Article CAS PubMed PubMed Central Google Scholar - Bawden, E. & Gebhardt, T. The multifaceted roles of CD4(+) T cells and MHC class II in cancer surveillance. Curr. Opin. Immunol. 83, 102345 (2023).
Article CAS PubMed Google Scholar - Richters, M. M. et al. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 11, 56 (2019).
Article PubMed PubMed Central Google Scholar - Fotakis, G., Trajanoski, Z. & Rieder, D. Computational cancer neoantigen prediction: current status and recent advances. Immunooncol. Technol. 12, 100052 (2021).
Article CAS PubMed PubMed Central Google Scholar - Vita, R. et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343 (2019).
Article CAS PubMed Google Scholar - Shao, W., Caron, E., Pedrioli, P. & Aebersold, R. The SysteMHC Atlas: a computational pipeline, a website, and a data repository for immunopeptidomic analyses. Methods Mol. Biol. 2120, 173–181 (2020).
Article CAS PubMed Google Scholar - Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Article CAS PubMed Google Scholar - Andreatta, M. & Nielsen, M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32, 511–517 (2016).
Article CAS PubMed Google Scholar - O’Donnell, T. J. et al. MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. 7, 129–132.e4 (2018).
Article PubMed Google Scholar - Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, W449–W454 (2020).
Article CAS PubMed PubMed Central Google Scholar - O’Donnell, T. J., Rubinsteyn, A. & Laserson, U. MHCflurry 2.0: improved pan-allele prediction of MHC class I-presented peptides by incorporating antigen processing. Cell Syst. 11, 418–419 (2020).
Article PubMed Google Scholar - Shim, J. H. et al. HLA-corrected tumor mutation burden and homologous recombination deficiency for the prediction of response to PD-(L)1 blockade in advanced non-small-cell lung cancer patients. Ann. Oncol. 31, 902–911 (2020).
Article CAS PubMed Google Scholar - Ghorani, E. et al. Differential binding affinity of mutated peptides for MHC class I is a predictor of survival in advanced lung cancer and melanoma. Ann. Oncol. 29, 271–279 (2018).
Article CAS PubMed Google Scholar - Balachandran, V. P. et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, 512–516 (2017).
Article CAS PubMed PubMed Central Google Scholar - Luksza, M. et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 551, 517–520 (2017).
Article CAS PubMed PubMed Central Google Scholar - Litchfield, K. et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 184, 596–614 e514 (2021).
Article CAS PubMed PubMed Central Google Scholar - Luksza, M. et al. Neoantigen quality predicts immunoediting in survivors of pancreatic cancer. Nature 606, 389–395 (2022).
Article CAS PubMed PubMed Central Google Scholar - Wells, D. K. et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell 183, 818–834.e13 (2020).
Article CAS PubMed PubMed Central Google Scholar - Rojas, L. A. et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 618, 144–150 (2023).
Article CAS PubMed PubMed Central Google Scholar - Weber, J. S. et al. Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): a randomised, phase 2b study. Lancet 403, 632–644 (2024).
Article CAS PubMed Google Scholar - Oreper, D., Klaeger, S., Jhunjhunwala, S. & Delamarre, L. The peptide woods are lovely, dark and deep: hunting for novel cancer antigens. Semin. Immunol. 67, 101758 (2023).
Article CAS PubMed Google Scholar - Ng, K. W. et al. Antibodies against endogenous retroviruses promote lung cancer immunotherapy. Nature 616, 563–573 (2023).
Article CAS PubMed PubMed Central Google Scholar - Panda, A. et al. Endogenous retrovirus expression is associated with response to immune checkpoint blockade in clear cell renal cell carcinoma. JCI Insight 3, e121522 (2018).
Article PubMed PubMed Central Google Scholar - McGranahan, N. et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016).
Article CAS PubMed PubMed Central Google Scholar - Wolf, Y. et al. UVB-induced tumor heterogeneity diminishes immune response in melanoma. Cell 179, 219–235.e21 (2019).
Article CAS PubMed PubMed Central Google Scholar - Boll, L. M. et al. The impact of mutational clonality in predicting the response to immune checkpoint inhibitors in advanced urothelial cancer. Sci. Rep. 13, 15287 (2023).
Article CAS PubMed PubMed Central Google Scholar - Westcott, P. M. K. et al. Mismatch repair deficiency is not sufficient to elicit tumor immunogenicity. Nat. Genet. 55, 1686–1695 (2023).
Article CAS PubMed PubMed Central Google Scholar - Ravi, A. et al. Genomic and transcriptomic analysis of checkpoint blockade response in advanced non-small cell lung cancer. Nat. Genet. 55, 807–819 (2023).
Article CAS PubMed PubMed Central Google Scholar - Freeman, S. S. et al. Combined tumor and immune signals from genomes or transcriptomes predict outcomes of checkpoint inhibition in melanoma. Cell Rep. Med. 3, 100500 (2022).
Article CAS PubMed PubMed Central Google Scholar - Niknafs, N. et al. Persistent mutation burden drives sustained anti-tumor immune responses. Nat. Med. 29, 440–449 (2023).
Article CAS PubMed PubMed Central Google Scholar - Anagnostou, V. et al. Integrative tumor and immune cell multi-omic analyses predict response to immune checkpoint blockade in melanoma. Cell Rep. Med. 1, 100139 (2020).
Article CAS PubMed PubMed Central Google Scholar - Markham, J. F. et al. Predicting response to immune checkpoint blockade in NSCLC with tumour-only RNA-seq. Br. J. Cancer 128, 1148–1154 (2023).
Article CAS PubMed Google Scholar - Szeto, C. et al. High correlation between TMB, expressed TMB, and neoantigen load using tumor: normal whole exome DNA and matched whole transcriptome RNA sequencing. J. Clin. Oncol. 38, e15238 (2020).
Article Google Scholar - DiGuardo, M. A. et al. RNA-seq reveals differences in expressed tumor mutation burden in colorectal and endometrial cancers with and without defective DNA-mismatch repair. J. Mol. Diagn. 23, 555–564 (2021).
Article CAS PubMed Google Scholar - Sorokin, M. et al. RNA sequencing data for FFPE tumor blocks can be used for robust estimation of tumor mutation burden in individual biosamples. Front. Oncol. 11, 732644 (2021).
Article CAS PubMed PubMed Central Google Scholar - Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Article CAS PubMed PubMed Central Google Scholar - Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Campbell, P. J. & Stratton, M. R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 3, 246–259 (2013).
Article CAS PubMed PubMed Central Google Scholar - Degasperi, A. et al. Substitution mutational signatures in whole-genome-sequenced cancers in the UK population. Science https://doi.org/10.1126/science.abl9283 (2022).
- Nik-Zainal, S. et al. Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 (2012).
Article CAS PubMed PubMed Central Google Scholar - Sha, D. et al. Tumor mutational burden as a predictive biomarker in solid tumors. Cancer Discov. 10, 1808–1825 (2020).
Article CAS PubMed PubMed Central Google Scholar - The COSMIC Database v3.4. https://cancer.sanger.ac.uk/signatures/ (2023).
- Zhao, E. Y. et al. Homologous recombination deficiency and platinum-based therapy outcomes in advanced breast cancer. Clin. Cancer Res. 23, 7521–7530 (2017).
Article CAS PubMed Google Scholar - Davies, H. et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 23, 517–525 (2017).
Article CAS PubMed PubMed Central Google Scholar - Nguyen, L., Martens, J. W. M., Van Hoeck, A. & Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 11, 5584 (2020).
Article CAS PubMed PubMed Central Google Scholar - Secrier, M. et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet. 48, 1131–1141 (2016).
Article CAS PubMed PubMed Central Google Scholar - Petrelli, A. et al. BRCA2 germline mutations identify gastric cancers responsive to PARP inhibitors. Cancer Res. 83, 1699–1710 (2023).
Article CAS PubMed PubMed Central Google Scholar - Peng, G. et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat. Commun. 5, 3361 (2014).
Article PubMed Google Scholar - Li, H. et al. PARP inhibitor resistance: the underlying mechanisms and clinical implications. Mol. Cancer 19, 107 (2020).
Article CAS PubMed PubMed Central Google Scholar - Le, D. T. et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520 (2015).
Article CAS PubMed PubMed Central Google Scholar - Le, D. T. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017).
Article CAS PubMed PubMed Central Google Scholar - Marabelle, A. et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 38, 1–10 (2020).
Article CAS PubMed Google Scholar - Touat, M. et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580, 517–523 (2020).
Article CAS PubMed PubMed Central Google Scholar - Chen, H. et al. The immune response-related mutational signatures and driver genes in non-small-cell lung cancer. Cancer Sci. 110, 2348–2356 (2019).
Article CAS PubMed PubMed Central Google Scholar - Miao, D. et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 50, 1271–1281 (2018).
Article CAS PubMed PubMed Central Google Scholar - Wang, S., Jia, M., He, Z. & Liu, X. S. APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene 37, 3924–3936 (2018).
Article CAS PubMed PubMed Central Google Scholar - Liao, J. et al. Clinical and genomic characterization of mutational signatures across human cancers. Int. J. Cancer 152, 1613–1629 (2023).
Article CAS PubMed Google Scholar - Chong, W. et al. Association of clock-like mutational signature with immune checkpoint inhibitor outcome in patients with melanoma and NSCLC. Mol. Ther. Nucleic Acids 23, 89–100 (2021).
Article CAS PubMed Google Scholar - Valero, C. et al. Clinical-genomic determinants of immune checkpoint blockade response in head and neck squamous cell carcinoma. J. Clin. Invest. 133, e169823 (2023).
Article CAS PubMed PubMed Central Google Scholar - Koh, G., Degasperi, A., Zou, X., Momen, S. & Nik-Zainal, S. Mutational signatures: 1267 emerging concepts, caveats and clinical applications. Nat. Rev. Cancer 21, 619–637 (2021).
Article CAS PubMed Google Scholar - NIH. The cost of sequencing a human genome. genome.gov https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost (2021).
- Newell, F. et al. Multiomic profiling of checkpoint inhibitor-treated melanoma: identifying predictors of response and resistance, and markers of biological discordance. Cancer Cell 40, 88–102.e7 (2022).
Article CAS PubMed Google Scholar - Liu, D. et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 25, 1916–1927 (2019).
Article CAS PubMed PubMed Central Google Scholar - Forde, P. M. et al. Durvalumab with platinum-pemetrexed for unresectable pleural mesothelioma: survival, genomic and immunologic analyses from the phase 2 PrE0505 trial. Nat. Med. 27, 1910–1920 (2021).
Article CAS PubMed PubMed Central Google Scholar - Anagnostou, V., Landon, B. V., Medina, J. E., Forde, P. & Velculescu, V. E. Translating the evolving molecular landscape of tumors to biomarkers of response for cancer immunotherapy. Sci. Transl. Med. 14, eabo3958 (2022).
Article CAS PubMed PubMed Central Google Scholar - Sinha, N. et al. Immune determinants of the association between tumor mutational burden and immunotherapy response across cancer types. Cancer Res. 82, 20762083 (2022).
Article Google Scholar - Danaher, P. et al. Gene expression markers of tumor infiltrating leukocytes. J. Immunother. Cancer 5, 18 (2017).
Article PubMed PubMed Central Google Scholar - Cristescu, R. et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362, eaar3593 (2018).
Article PubMed PubMed Central Google Scholar - Goodman, A. M. et al. MHC-I genotype and tumor mutational burden predict response to immunotherapy. Genome Med. 12, 45 (2020).
Article CAS PubMed PubMed Central Google Scholar - Han, J. et al. Pan-cancer analysis reveals sex-specific signatures in the tumor microenvironment. Mol. Oncol. 16, 2153–2173 (2022).
Article CAS PubMed PubMed Central Google Scholar - Lee, J. S. & Ruppin, E. Multiomics prediction of response rates to therapies to inhibit programmed cell death 1 and programmed cell death 1 ligand 1. JAMA Oncol. 5, 1614–1618 (2019).
Article CAS PubMed PubMed Central Google Scholar - Stenzinger, A., Kazdal, D. & Peters, S. Strength in numbers: predicting response to checkpoint inhibitors from large clinical datasets. Cell 184, 571–573 (2021).
Article CAS PubMed Google Scholar - Mason, M. et al. A community challenge to predict clinical outcomes after immune checkpoint blockade in non-small cell lung cancer. J. Transl. Med. 22, 190 (2024).
Article PubMed PubMed Central Google Scholar - Gajic, Z. Z., Deshpande, A., Legut, M., Imielinski, M. & Sanjana, N. E. Recurrent somatic mutations as predictors of immunotherapy response. Nat. Commun. 13, 3938 (2022).
Article CAS PubMed PubMed Central Google Scholar - Wang, J. et al. Mutational analysis of microsatellite-stable gastrointestinal cancer with high tumour mutational burden: a retrospective cohort study. Lancet Oncol. 24, 151–161 (2023).
Article PubMed PubMed Central Google Scholar - Colle, R. et al. BRAF V600E/RAS mutations and Lynch syndrome in patients with MSIH/dMMR metastatic colorectal cancer treated with immune checkpoint inhibitors. Oncologist 28, 771–779 (2023).
Article PubMed PubMed Central Google Scholar - Liu, G. C. et al. The heterogeneity between Lynch-associated and sporadic MMR deficiency in colorectal cancers. J. Natl Cancer Inst. 110, 975–984 (2018).
Article PubMed Google Scholar - Ratovomanana, T. et al. Prediction of response to immune checkpoint blockade in patients with metastatic colorectal cancer with microsatellite instability. Ann. Oncol. 34, 703–713 (2023).
Article CAS PubMed Google Scholar - Zhang, J. et al. The combination of neoantigen quality and T lymphocyte infiltrates identifies glioblastomas with the longest survival. Commun. Biol. 2, 135 (2019).
Article PubMed PubMed Central Google Scholar - Sade-Feldman, M. et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 8, 1136 (2017).
Article PubMed PubMed Central Google Scholar - Sucker, A. et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin. Cancer Res. 20, 6593–6604 (2014).
Article CAS PubMed PubMed Central Google Scholar - Mumphrey, M. B. et al. Distinct mutational processes shape selection of MHC class I and class II mutations across primary and metastatic tumors. Cell Rep. 42, 112965 (2023).
Article CAS PubMed Google Scholar - Middha, S. et al. Majority of B2M-mutant and -deficient colorectal carcinomas achieve clinical benefit from immune checkpoint inhibitor therapy and are microsatellite instability-high. JCO Precis. Oncol. https://doi.org/10.1200/PO.18.00321 (2019).
- Tikidzhieva, A. et al. Microsatellite instability and beta2-microglobulin mutations as prognostic markers in colon cancer: results of the FOGT-4 trial. Br. J. Cancer 106, 12391245 (2012).
Article Google Scholar - Barrow, P. et al. Confirmation that somatic mutations of beta-2 microglobulin correlate with a lack of recurrence in a subset of stage II mismatch repair deficient colorectal cancers from the QUASAR trial. Histopathology 75, 236–246 (2019).
Article PubMed PubMed Central Google Scholar - Busch, E. et al. Beta-2-microglobulin mutations are linked to a distinct metastatic pattern and a favorable outcome in microsatellite-unstable stage IV gastrointestinal cancers. Front. Oncol. 11, 669774 (2021).
Article CAS PubMed PubMed Central Google Scholar - Germano, G. et al. CD4 T cell-dependent rejection of beta-2 microglobulin null mismatch repair-deficient tumors. Cancer Discov. 11, 1844–1859 (2021).
Article CAS PubMed Google Scholar - de Vries, N. L. et al. γδ T cells are effectors of immunotherapy in cancers with HLA class I defects. Nature 613, 743–750 (2023).
Article PubMed PubMed Central Google Scholar - Marabelle, A., Aspeslagh, S., Postel-Vinay, S. & Soria, J. C. JAK mutations as escape mechanisms to anti-PD-1 therapy. Cancer Discov. 7, 128–130 (2017).
Article CAS PubMed Google Scholar - Bayle, A. et al. ESMO study on the availability and accessibility of biomolecular technologies in oncology in Europe. Ann. Oncol. 34, 934–945 (2023).
Article CAS PubMed Google Scholar - Genomics England. The 100,000 Genomes Project. https://www.genomicsengland.co.uk/initiatives/100000-genomes-project (2024).
- The German Federal Ministry of Health. GenomeDE — National Strategy for Genomic Medicine. https://www.bundesgesundheitsministerium.de/en/en/international/european-health-policy/genomde-en.html (2024).
- The US National Cancer Institute Pan-Cancer Atlas. https://gdc.cancer.gov/about-data/publications/pancanatlas (2024).
- Maruvka, Y. E. et al. Analysis of somatic microsatellite indels identifies driver events in human tumors. Nat. Biotechnol. 35, 951–959 (2017).
Article CAS PubMed PubMed Central Google Scholar - Marcus, L. et al. FDA approval summary: pembrolizumab for the treatment of tumor mutational burden-high solid tumors. Clin. Cancer Res. 27, 4685–4689 (2021).
Article CAS PubMed PubMed Central Google Scholar
Author information
Author notes
- These authors contributed equally: Jan Budczies, Daniel Kazdal.
Authors and Affiliations
- Institute of Pathology, Heidelberg University Hospital, Heidelberg, Germany
Jan Budczies, Daniel Kazdal, Michael Menzel, Susanne Beck, Klaus Kluck, Christian Altbürger, Constantin Schwab, Michael Allgäuer, Peter Schirmacher & Albrecht Stenzinger - Translational Lung Research Center (TLRC) Heidelberg, Member of the German Center for Lung Research (DZL), Heidelberg, Germany
Jan Budczies, Daniel Kazdal, Petros Christopoulos & Albrecht Stenzinger - Center for Personalized Medicine (ZPM), Heidelberg, Germany
Jan Budczies, Daniel Kazdal, Michael Menzel, Susanne Beck, Klaus Kluck, Christian Altbürger, Constantin Schwab, Michael Allgäuer, Peter Schirmacher & Albrecht Stenzinger - Department of Applied Tumour Biology, Institute of Pathology, Heidelberg University Hospital, Heidelberg, Germany
Aysel Ahadova & Matthias Kloor - Clinical Cooperation Unit Applied Tumour Biology, German Cancer Research Center (DKFZ), Heidelberg, Germany
Aysel Ahadova & Matthias Kloor - Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne University, Lausanne, Switzerland
Solange Peters - Clinical Cooperation Unit Molecular Hematology/Oncology, German Cancer Research Center (DKFZ), Heidelberg, Germany
Alwin Krämer - Department of Internal Medicine V, University of Heidelberg, Heidelberg, Germany
Alwin Krämer - Department of Thoracic Oncology, Thoraxklinik and National Center for Tumour Diseases at Heidelberg University Hospital, Heidelberg, Germany
Petros Christopoulos
Authors
- Jan Budczies
You can also search for this author inPubMed Google Scholar - Daniel Kazdal
You can also search for this author inPubMed Google Scholar - Michael Menzel
You can also search for this author inPubMed Google Scholar - Susanne Beck
You can also search for this author inPubMed Google Scholar - Klaus Kluck
You can also search for this author inPubMed Google Scholar - Christian Altbürger
You can also search for this author inPubMed Google Scholar - Constantin Schwab
You can also search for this author inPubMed Google Scholar - Michael Allgäuer
You can also search for this author inPubMed Google Scholar - Aysel Ahadova
You can also search for this author inPubMed Google Scholar - Matthias Kloor
You can also search for this author inPubMed Google Scholar - Peter Schirmacher
You can also search for this author inPubMed Google Scholar - Solange Peters
You can also search for this author inPubMed Google Scholar - Alwin Krämer
You can also search for this author inPubMed Google Scholar - Petros Christopoulos
You can also search for this author inPubMed Google Scholar - Albrecht Stenzinger
You can also search for this author inPubMed Google Scholar
Contributions
J.B., M.M., S.B., K.K., C.A., C.S., A.A., M.K. and A.K. researched data for the manuscript; J.B., D.K., M.M., S.B., K.K., P.S., S.P. and A.S. made a substantial contribution to discussions of content; J.B., D.K., C.A., C.S., A.A., M.K., A.K., P.C. and A.S. wrote the manuscript; and all authors reviewed and/or edited before submission.
Corresponding authors
Correspondence toJan Budczies or Albrecht Stenzinger.
Ethics declarations
Competing interests
J.B. has acted as a consultant of MSD. D.K. has acted as a consultant and/or adviser of Agilent, AstraZeneca, BMS, Illumina, Incyte, Eli Lilly, Pfizer and Takeda. C.S. has acted as a consultant of MSD and has received speaker’s fees from Bayer Vital and Boehringer Ingelheim. P.S. has acted as a consultant of BMS, Eisai, Incyte, Janssen, MSD and Roche and has received research funding from BMS, Chugai and Incyte. S.P. has acted as a consultant and/or adviser of AbbVie, Amgen, Arcus, AstraZeneca, Bayer, Beigene, BerGenBio, Biocartis, BioInvent, Blueprint Medicines, Boehringer Ingelheim, Bristol-Myers Squibb, Clovis, Daiichi Sankyo, Debiopharm, Eli Lilly, F-Star, Fishawack, Foundation Medicine, Genzyme, Gilead, GlaxoSmithKline, Hutchmed, Illumina, Incyte, Ipsen, iTeos, Janssen, Merck Sharp & Dohme, Merck Serono, Merrimack, Mirati, Nykode Therapeutics, Novartis, Novocure, Pharma Mar, Promontory Therapeutics, Pfizer, Regeneron, Roche/Genentech, Sanofi, Seattle Genetics and Takeda; holds a board of director position for Galenica; has acted as a speaker for AstraZeneca, Boehringer Ingelheim, BMS, Eli Lilly, Foundation Medicine, GlaxoSmithKline, Illumina, Ipsen, Merck Sharp & Dohme, Mirati, Novartis, Pfizer, Roche/Genentech, Sanofi and Takeda; and has acted as a principal investigator in trials sponsored by Amgen, Arcus, AstraZeneca, Beigene, Bristol-Myers Squibb, GlaxoSmithKline, iTeos, Merck Sharp & Dohme, Mirati, Pharma Mar, Promontory Therapeutics, Roche/Genentech and Seattle Genetics. A.K. has acted as a consultant and/or adviser, received support for attending meetings and/or travel, participated in a data safety monitoring board and received research funding from F. Hoffmann-La Roche and has received research funding from BMS and Molecular Health. P.C. has acted as a consultant and/or adviser of AstraZeneca, Boehringer Ingelheim, Chugai, Pfizer, Novartis, MSD, Takeda and Roche; has acted as a speaker for AstraZeneca, Janssen, Novartis, Roche, Pfizer, Thermo Fisher and Takeda; has received research funding from AstraZeneca, Amgen, Boehringer Ingelheim, Novartis, Roche and Takeda; and has received travel support from AstraZeneca, Eli Lilly, Daiichi Sankyo, Gilead, Novartis, Pfizer and Takeda. A.S. has acted as an adviser and/or speaker for Agilent, Aignostics, Amgen, Astellas, AstraZeneca, Bayer, BMS, Eli Lilly, Illumina, Incyte, Janssen, MSD, Novartis, Pfizer, Qlucore, QuIP, Roche, Sanofi, Seagen, Servier, Takeda and Thermo Fisher Scientific and has received research funding from Bayer, BMS, Chugai, Incyte and MSD.
Peer review
Peer review information
Nature Reviews Clinical Oncology thanks A. Schoenfeld, R. Thummalapalli and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article
Cite this article
Budczies, J., Kazdal, D., Menzel, M. et al. Tumour mutational burden: clinical utility, challenges and emerging improvements.Nat Rev Clin Oncol 21, 725–742 (2024). https://doi.org/10.1038/s41571-024-00932-9
- Accepted: 23 July 2024
- Published: 27 August 2024
- Issue Date: October 2024
- DOI: https://doi.org/10.1038/s41571-024-00932-9