Role of the nuclear envelope on genome organization and gene expression (original) (raw)
. Author manuscript; available in PMC: 2012 Mar 1.
Published in final edited form as: Wiley Interdiscip Rev Syst Biol Med. 2011 Mar;3(2):147–166. doi: 10.1002/wsbm.101
While often depicted as a static structure upon which proteinaceous factors bind to control gene expression, the genome is actually highly mobile and capable of exploring the complex domain architecture of the nucleus, which in turn controls genome maintenance and gene expression. Numerous genes relocate from the nuclear periphery to the nuclear interior upon activation and are hypothesized to interact with pre-assembled sites of transcription. In contrast to the nuclear interior, the nuclear periphery is widely regarded as transcriptionally silent. This is reflected by the preferential association of heterochromatin with the nuclear envelope. However, some activated genes are recruited to the nuclear periphery through interactions with nuclear pore complexes (NPCs) and NPC components are capable of preventing the spread of silent chromatin into adjacent regions of active chromatin, leading to the speculation that NPCs may facilitate the transition of chromatin between transcriptional states. Thus, the nuclear envelope (NE) might better be considered as a discontinuous platform that promotes both gene activation and repression. As such, it is perhaps not surprising that many disease states are frequently associated with alterations in the NE. Here we review the effects of the nuclear envelope and its constituents on chromatin organization and gene expression.
While genome sequencing continues to enumerate the genomic potential of a multitude of organisms, understanding what genes are expressed, when they are expressed and how they are expressed are fundamental challenges for biology. But because, at any given time, all genes in a genome do not have the same potential to be expressed, predicting gene expression is not simply a matter of knowing what transcription factors are present and where they bind. As early as 1928, Emil Heitz noted at least two types of chromatin: highly condensed chromatin that failed to decondense during interphase and other regions on the same chromosome that did.1, 2 He referred to these distinct chromosomal domains as heterochromatin and euchromatin, respectively. Eighty years later we know that DNA must be highly organized and packed for it to even fit in the nucleus of a cell and that tightly packed regions of chromatin are generally transcriptionally silent, whereas loosely packed chromatin has greater potential to be transcriptionally active. DNA organization of this sort is accomplished through multiple layers of compaction to form chromatin. The first order of compaction consists of 146 base pairs of DNA wound around an octamer of histone proteins to form a nucleosome. Nucleosomes oligomerize to form a 30nm fiber and ultimately organize into a poorly defined higher order chromatin structure. Each layer of compaction is highly regulated and subtle changes induced by chromatin binding proteins and histone modifying enzymes can greatly influence the accessibility of DNA to transcription factors, leading to regions that are transcriptionally active or repressed, as is the case for Emil Heitz's euchromatin and heterochromatin, respectively (reviewed in3, 4). The N-termini of histones are extensively post-translationally modified by acetylation, methylation, phosphorylation, ubiquitination or sumoylation. Hyperacetylation of lysine residues is correlated with increased transcription, particularly acetylation of lysine 16 on histone H4 (H4 K16ac) whereas lysine hypoacetylation and methylation of lysine 9 on histone H3 (H3 K9me) are hallmarks of heterochromatin (reviewed in5, 6). To prevent the spread of one chromatin state into an adjacent region of the opposite chromatin state, boundary regions are present that lie at the interfaces of opposing chromatin. Such boundary regions are often dynamic and contain epigenetic marks of both active and inactive chromatin.7-9
Early electron micrographs revealed that although these highly condensed chromatin domains are found both at the nuclear periphery and nuclear interior, for many cell types they are preferentially localized to the nuclear periphery, suggesting, that regions interior to the nucleus tend to promote gene expression, while regions at the periphery tend to promote gene silencing. An exception to this generality (which has been evident since these early images) is near nuclear pore complexes – complex macromolecular structures embedded in the nuclear envelope that permit and regulate macromolecular transport between the nucleus and the cytoplasm. While these morphological characteristics have been a source of speculation regarding gene expression10, 11, the molecular mechanisms and functional impact of the three-dimensional nuclear architecture on gene expression is only now beginning to be understood.
The nuclear envelope (NE) is a double-membrane system surrounding the nuclear chromatin. It is comprised of an outer nuclear membrane, which is continuous with the endoplasmic reticulum, and an inner nuclear membrane exposed to the nucleoplasm. Underlying the inner nuclear membrane in metazoan cells is the nuclear lamina, a complex meshwork of intermediate filaments comprised of lamin A, C, B1 and B2, which connect to the NE through multiple integral inner membrane proteins (reviewed in12-15). The primary function of the lamina has been thought to maintain nuclear shape in concert with the NE, however in recent years it has become clear that the lamina has important functions in transcriptional regulation and genome organization.12, 16, 17 Mutations that affect the functions of lamins often lead to a diverse set of tissue specific diseases referred to as laminopathies, which include forms of muscular dystrophy, lipodystrophy and premature ageing syndromes (reviewed in18-22).
Approximately 30 different nuclear pore proteins (nucleoporins or nups) comprise each of the numerous, large, and proteinaceous nuclear pore complexes (NPCs) that are distributed throughout the nuclear envelope, thereby providing gateways for exchange between the nucleoplasm and cytoplasm. Each NPC is highly modular and symmetric in structure, forming an octagonal donut-like structure embedded in, and spanning the double membrane of the NE. Consistent with the morphological symmetry, each nucleoporin is present in multiple (at least 8) copies.23 While the majority of nucleoporins are symmetrically located on both the cytoplasmic and nucleoplasmic sides of the NPC, there are those that are localized specifically to either the cytoplasmic or nucleoplasmic face of the NPC. On the cytoplasmic side, these nucleoporins (e.g. Nup159p and Nup82p in yeast or Nup214 and Nup88 in mammals) form flexible filaments that emanate into the cytoplasm apparently like tentacles of an octopus. On the nucleoplasmic side, (Nup1p, Nup2p and Nup60p in yeast or Nup50, and Nup153 in mammals) and associated proteins (Mlp1p and Mlp2p in yeast or Tpr in mammals) form a basket-like structure that can extend upwards of ∼100 nm into the nucleus and is situated in a prime location to facilitate interactions with underlying chromatin (Figure 1).24, 25
FIGURE 1.
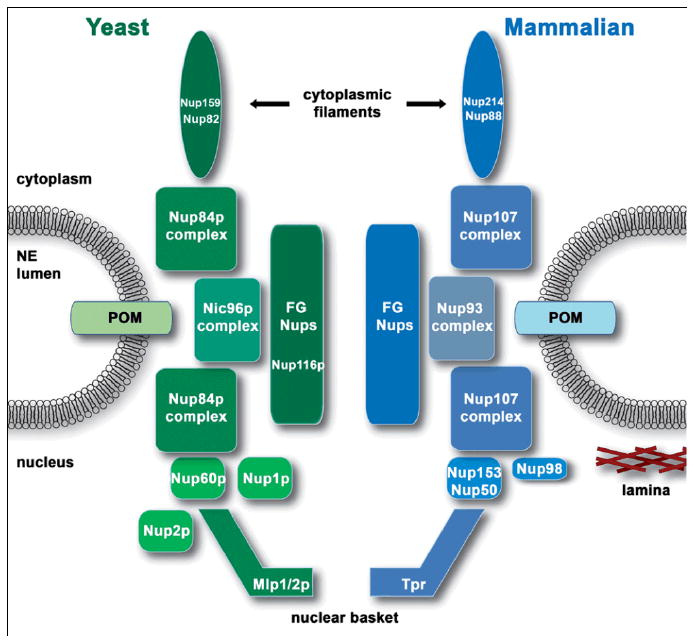
Nuclear pore complex. The NPC is a large proteinaceous structure that extends across the nuclear envelope at points where the inner and outer nuclear membranes are fused, creating points of transit between the nucleoplasm and cytoplasm. Interactions with FG-nups lining the central channel facilitates transport of import and export complexes through the NPC. Major sub-complexes of the yeast NPC are shown in green (left side of image) and the corresponding mammalian homologs are shown in blue (right side of image). Positions of specific nucleoporins discussed throughout the text as having roles in gene expression are depicted. POM, pore outer membrane (integral NE proteins that anchor NPCs to the NE; nup, nucleoporin; NE, nuclear envelope; Mlp, myosin like protein; Tpr, translocated promoter region; FG-nups, nucleoporins rich in phenylalanine-glycine repeats.
Consistent with early morphological observations, localization of specific chromosomal regions within the nucleus revealed that transcriptionally inactive regions such as telomeres, centromeres, the inactive X chromosome and in yeast, the stably repressed mating type loci (HMR and HML) are preferentially associated with the nuclear periphery. Moreover, genes activated during cellular differentiation often translocate away from the periphery towards the nuclear interior, as is the case for immunoglobulin loci during B-cell activation.26, 27 However, the nuclear periphery can also promote transcription as demonstrated by numerous yeast genes that are recruited to the nuclear periphery for full transcriptional activity.28-34 The mechanism(s) that determine whether a peripherally localized gene is active or repressed are not clear, but likely depend on the context of the surrounding nuclear architecture including interactions with specific subdomains of the NE, contacts with the nuclear lamina in metazoans and the state of the surrounding chromatin.
How are these epigenetic states (and expression) of the chromatin influenced by the three dimensional architecture of the nucleus? Considering the multitude of parameters influencing the epigenetic states, it seems impractical to think we can generalize what is learned from single genes to the rest of the genome. One promising approach that is beginning to reveal principles governing gene expression involves systems biology (Box 1). In such an approach, we can consider the influence of directed perturbations on a genome-wide scale. We can also begin to consider an integrated approach involving global interrogations of the dynamics of protein-DNA interactions, molecular epigenetic marks on chromosomes, expression states etc., and to explore these data computationally to build models of function that can begin to explain how the structural dynamics of the genome and the cell influence and control expression of the cell's blueprint.
BOX 1. Systems biology approaches to study the function of NPCs in gene expression.
Systems biology has emerged as an important field in understanding complex biological systems. The power of systems biology lies in its ability to integrate high-throughput genetics, functional genomics and proteomics with computational data analyses to identify associations and patterns within complex data sets. Systems biology has been successfully used to understand the function of NPCs in gene regulation. Analysis of global mRNA levels using microarray-based techniques identified the boundary activity of endogenous NPCs.57 ChIP-CHIP approaches identified specific nuclear pore-promoter interactions, which were further characterized as an early physiological event in transcriptional activation. Furthermore, proteomic methods have been successfully applied to identify protein complexes involved in establishment and maintenance of chromatin boundaries.89 Recently, computational genomics discovered over 97 promoters with perfect sequence identity to the gene recruitment sequence (GRS) of the INO1 gene, which functions to target INO1 to the nuclear periphery during activation.78
Nuclear envelope associated heterochromatin
Role of the nuclear envelope in metazoan differentiation
Advances in microscopy techniques have revealed the dynamic nature of the chromosomal domains. In-vivo visualization and real-time tracking of discrete chromosomal loci (Figure 2) in mammalian cells have led to the discovery that chromosomes encoding relatively few genes, said to be gene-poor, are more frequently associated with the nuclear periphery whereas gene-rich chromosomes are positioned more internally.35, 36 Similarly, highly transcribed, co-regulated genes often cluster within chromosomal territories, as is the case for the murine immunoglobulin gene clusters Igh and Igk, encoding genes of the immunoglobulin heavy and light chains respectively, which, in hematopoietic progenitor B-cells, associate with the nuclear lamina when inactive and relocate away from the nuclear periphery following commitment to the B-cell lineage.26, 27 In this context, peripheral localization is proposed to prevent spurious immunoglobulin rearrangements by limiting accessibility of Ig loci to recombination factors.37 Similar gene repositioning occurs with the MASH1 locus during neural induction of embryonic stem cells and the beta-globin genes during erythrocyte maturation.38, 39 NANOG also undergoes gene repositioning during cell differentiation; however, in this case, because NANOG expression is required to maintain pluripotency of human embryonic stem cells, upon differentiation NANOG becomes transcriptionally silent and relocates from the nuclear interior to a more peripheral position.40 These observations correlate changes in gene positioning with changes in transcriptional activity during cell differentiation, and raise the question of whether gene relocalization is the cause or consequence of gene activation.
FIGURE 2.
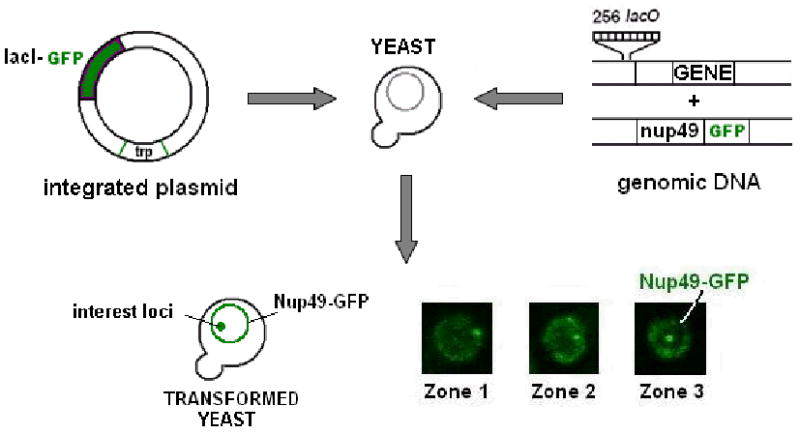
Chromosome tagging. The bacterial lac operator (LacO) and lac repressor (LacI) system in combination with green fluorescent protein (GFP) has been adapted for visualization of genome organization and dynamics in living cells.137 The system depends on high expression levels of LacI-GFP, encoded either ectopically on a plasmid or genomically integrated, in conjunction with integration of a tandem array of 256 LacO sequences at the desired genomic locus. LacO sequences are tightly bound by LacI-GFP and are visualized as a bright green focus by fluorescence microscopy (central foci in Zone 3; lower right panel). High expression levels of LacI-GFP bound in relatively close proximity provide a focus that is easily observed with minimal excitation and can be repeatedly visualized with minimal photobleaching, making it ideal for time lapse microscopy in living cells. The addition of a fluorescent marker of the nuclear periphery such as a GFP tagged nucleoporin, Nup49-GFP, and division of the nucleus into three concentric zones of equal area, zone 1-3, aid in determining gene positioning. Chromosome tagging has been successfully used to follow gene loci in both yeast and mammalian systems and has been adapted as a tool to tether genes to the nuclear envelope through the fusion of LacI to an integral inner nuclear membrane protein.29, 37
Silent domains at the nuclear periphery: lessons from yeast
Although budding yeast lack the hallmarks of heterochromatin found in higher eukaryotes such as H3 K9me and the compaction required to visualize heterochromatin by microscopy, silencing nonetheless occurs in heterochromatin-like regions and this model organism has been instrumental to our understanding of the importance of gene positioning in transcriptional regulation. Silent regions in yeast including centromeres, telomeres and the silent mating type loci (HMR and HML), all localize in the vicinity of the nuclear periphery.41-43 Yeast telomeres are assembled into silent chromatin and coalesce to form clusters of 8-10 foci per cell.44-46 These telomeric foci are tethered at the periphery through two redundant mechanisms involving interactions between Sir4p, a component of the silencing machinery, and the peripheral inner nuclear membrane protein, Esc1p or between the DNA-end binding yKu heterodimer and the integral inner nuclear membrane protein Mps3p (reviewed in47-49). Combinatory deletions that negate both pathways lead to dispersal of telomere foci into the nuclear interior.50 Microarray analysis revealed a loss of subtelomeric silencing concomitant with decreased expression of non-telomeric genes, a result attributed to dispersal to ectopic chromatin sites of the silent information regulatory complex (Sir2p-Sir3p-Sir4p) that is required for silencing.50 Therefore, it was concluded that telomere clustering at the periphery sequesters a limited pool of Sir proteins at concentrations high enough to permit heterochromatin formation. In support of this, silencing of an HMR locus flanked by weakened silencing elements is enhanced when artificially tethered to the nuclear periphery and enhanced silencing is dependent on intact peripheral telomere foci.51 However, peripheral tethering of an HMR locus lacking silencing elements failed to repress, suggesting that a peripheral localization promotes but is not sufficient for repression.
Nuclear lamina is linked to chromatin and gene expression
In higher eukaryotes, a mesh-like network lies directly beneath the nuclear envelope. This nuclear lamina structure is comprised of intermediate filament proteins called lamins, which are anchored to the inner nuclear membrane through associations with integral membrane proteins or through membrane insertion of a lipid moiety. The nuclear lamina is positioned to be a key regulator of gene expression through physical organization of the genome or by acting as a scaffold for dynamic interplay of effectors with the genome. Accordingly, in vitro binding studies suggest that lamins physically interact with the genome either through binding of histones or by directly binding to specific DNA sequences termed matrix attachment regions (MARs).52, 53 The first in vivo high resolution mapping of interactions between the genome and the nuclear lamina were performed in the fly Drosophila melanogaster using a genome-wide approach termed DamID (Box 2).54 In brief, the Escherichia coli adenine methyltransferase was fused to the B-type lamin (Lam) to generate a lamin chimera with DNA methlylation activity. The chimera was expressed in vivo and DNA that came in physical proximity to the B-type lamin was methylated on adenine residues. Subsequent specific isolation of methylated adenine DNA fragments followed by amplification, labeling and hybridization to microarrays resulted in the identification of 474 genes organized into 52 gene clusters that are targets of Lam binding. Microscopy analysis to determine nuclear gene positioning confirmed that Lam target genes were more likely to have a peripheral localization than non-Lam target genes. Importantly, genome wide transcriptional profiling suggested that Lam target genes are expressed at lower levels than non-target genes and chromatin immunoprecipitation revealed that lamin-associated chromatin is reduced in the histone modifications H3 K4me3 and H4 K16ac, which are associated with active genes.54 Furthermore, upon induction of differentiation the expression level of genes newly associated with Lam decreased, and expression of genes that lost Lam association increased, consistent with the hypothesis that transcriptionally inactive genes associate with lamins at the nuclear periphery. Surprisingly, further ChIP experiments showed that (at least in flies) the Lam-associated transcriptionally inactive genes adopt a chromatin state distinct from ‘classical’ heterochromatin. Specifically, heterochromatin markers, heterochromatin protein 1 (HP1) and Su(var)3-9 (recently renamed, Drosophila lysine methyltransferase 1 (dKMT1) were not found at Lam target genes.
BOX 2. Identification of genomic lamin binding sites by DamID.
A novel protein-genome interaction mapping technology termed “DamID” is based on the construction of a fusion protein consisting of the Escherichia coli DNA adenine methyltransferase (Dam) and a chromatin binding protein of interest.175 Binding of the Dam-fusion to genomic sites permits Dam-mediated adenine methylation of adjacent DNA. DNA isolation, followed by specific amplification of adenine methylated DNA fragments, permits identification of the amplified regions by either direct sequencing or microarray analysis. DamID has been successfully used in Drosophila and mammalian systems to assemble genome-wide profiles of DNA sequences bound to the nuclear lamina.37, 54 These data further demonstrated that transcriptionally inactive genes associate with the lamina.
What are the determinants of Lam binding? The Fornerod laboratory correlated Lam binding with regions of late replication, low transcriptional levels and an absence of active histone marks.54 A histone deacetylase inhibitor reduced global Lam association, consistent with the finding that Lam binding sites are often flanked by long, intergenic hypoacetylated regions, suggesting that hypoacetylation promotes interaction with the lamina. However, the histone deacetylase HDAC3 also binds to the nuclear periphery, through the lamin associated protein Lap2β in mammalian cells.55 Thus, raising a familiar question: Are the chromatin characteristics of Lam binding sites the cause or consequence of Lam association? Peripheral HDAC3 activity could be required to form hypoacetylated chromatin thereby promoting Lam binding, or alternatively, hypoacetylation and gene inactivation may occur as a consequence of chromatin recruitment to the nuclear periphery through association with Lam. In the latter case, Lam-association may be stablized by hypoacetylation, establishing local feedback.
Nuclear pore complexes: gene activation and chromatin boundaries
Activated genes associate with nuclear pore complexes
Over the last decade it has become increasingly evident that highly transcribed genes associate with nuclear pore complex proteins in yeast. Genome-wide chromosome immunopreciptation studies (ChIP-chip) have been performed on several components of the nuclear pore complex as well as multiple karyopherins, - the soluble proteins that mediate nuclear transport.56 These studies revealed that the NPC components Nup2p, Nup60p, Nup166p, Nic96p, Mlp1p and Mlp2p and the karyopherins Cse1p, Kap95p and Xpo1p preferentially associate with actively transcribed genes, enriching with genes that are highly transcribed such as those involved in glycolysis and ribosome biogenesis. In contrast the Ran guanine-nucleotide exchange factor (RanGEF), Prp20p, which is mechanistically and physically linked to the dynamics of the nuclear import and export cycle, preferentially associates with inactive genes.56 Prp20p is an essential transport factor that promotes the dissociation of karyopherin-cargo import complexes in the nucleus and associates with chromatin.57-59 Prp20p association with inactive genes has been proposed to target newly imported transcription factors directly to target gene promoters.56 Under repressive conditions the galactose-inducible GAL genes associate with Prp20p, but not with nucleoporins. Following the addition of galactose, the GAL genes associate with nucleoporins, but no longer associate with Prp20p, suggesting that a subset of genes can be recruited to the NPC under conditions of transcriptional activation.56, 60
In a different approach to identify genomic binding sites of nucleoporins, a chimera was generated between Nup2p and micrococcal nuclease (MN) whereby the nuclease activity of the chimeric protein can be activated by the addition of calcium ions (Box 3).61 Upon expression of the chimera in yeast and subsequent activation of the nuclease by calcium addition, the chromatin in the vicinity of the Nup2-MN chimera (and presumably in the vicinity of Nup2p under normal conditions) is cleaved. Mapping of the double-stranded breaks allowed identification of Nup2p binding sites within promoter-proximal regions, referred to as Nup-promoter interactions (Nup-PIs). In the case of the GAL genes, Nup-PI appears to be an early event in gene activation as association with the promoter region required induction by galactose and the transcriptional activator Gal4p, yet occurred independently of recruitment of the SAGA complex or RNAPII. These observations point to a complex network of dynamic interactions that exist between actively transcribed genes and nucleoporins, presumably reflecting the association of these genes with the nuclear pore complex. It should be stressed however, that although many active genes have been identified in association with NPCs, interaction with the nuclear periphery is not a mandatory feature of gene activity in yeast.62 Some genes appear targeted to the periphery independent of transcription63, not all highly expressed genes associate with the periphery and it seems unlikely that genes would associate stably with the nuclear pore complex itself during expression. There are certainly dynamics, gene specific parameters and accompanying chromatin and trancriptional regulators involved and the mechanisms are yet to be revealed.
BOX 3. Identification of nuclear pore complex-promoter interactions by ChEC.
Chromatin immunoprecipitation (ChIP) has been widely applied to identify protein-chromatin interactions. An alternative, complementary technique suggested to have greater resolution and sensitivity than ChIP is chromatin endogenous cleavage (ChEC).176 ChEC uses a chimeric protein consisting of microccocal nuclease (MN) fused to a chromatin binding protein of interest to target associated DNA for cleavage. When expressed in cells, MN fusion protein remains inactive. However, following isolation of cell nuclei, calcium is added to induce nuclease activity resulting in double-strand DNA breaks in the vicinity of the MN-fusion protein binding site. Isolation of DNA and mapping of dsDNA breaks permits identification of cleavage sites and the corresponding binding sites of the MN-fusion protein. Schmid et al. 2006 successfully applied ChEC to map the binding sites of a MN-tagged fusion of the nuclear pore protein Nup2p and determined preferential binding of Nup2p to promoter-proximal regions.61
Nevertheless, chromatin association with the NPC is a feature of higher organisms as well as yeast. ChIP studies of Nup93 revealed association with transcriptionally inactive regions and chromatin domains bearing histone modifications indicative of heterochromatin.64 Inhibition of histone deacetylases (HDACs) promotes global changes in transcription and the promoters of genes that increase in expression, also become associated with Nup93. In Drosophila, the hyperactivation of the X-chromosome in males is associated with peripheral localization and NPC association65, 66 and the HSP70 locus associates with the NPC and localizes at the nuclear periphery.67 Most recently, two papers have reported nucleoporin contacts with both silent loci and developmentally regulated genes undergoing transcriptional induction in Drosophila melanogaster.68, 69 However, like yeast Nup2p, some Drosophila nups are highly dynamic and exchange between NPCs and the nucleoplasm, and these studies demonstrated that soluble intranuclear pools of some nups can interact with chromatin. Thus nup-chromatin interactions are not necessarily restricted to the nuclear periphery. These observations provide an important advance in our understanding of the basic roles of nucleoporins in gene regulation of higher eukaryotes and suggest that nucleoplasmic nups could play an active role in gene regulation, perhaps as direct regulators, or as scaffolds to bring modifiers, transcription factors and the genome in proximity. Nonetheless, given the complexities of gene regulatory mechanisms governing development, it is reasonable to assume that remodeling of NPC-chromatin contacts could represent yet another layer of complexity on the modulation of gene expression.
In yeast, visualization of inducible genes (INO1, HXK1, GAL1, GAL2, SUC2, FIG2 and HSP104) through chromatin tagging confirmed that these loci relocate to the nuclear periphery and nuclear pore complexes upon transcriptional activation.28-30, 32, 56 However, the molecular mechanisms and functional significance of recruitment are not well understood. What factors mediate gene recruitment to NPCs? Several laboratories have sought to address this question and have uncovered a diverse set of factors that contribute to chromatin-NPC interactions including transcription factors, chromatin modifying complexes, a histone variant, and mRNA processing and export factors.28, 29, 32, 61, 65, 70-73 With respect to the NPC, it seems the nuclear basket proteins are important for gene recruitment. Nup2p binds the nuclear basket through Nup60p, interacts with chromatin through association with chromatin-bound Prp20p and is required for peripheral gene recruitment of GAL1 and INO1.57, 63, 74, 75 Also anchored to the nuclear basket via Nup60p are the myosin like proteins Mlp1p and Mlp2p that form coiled-coils that extend beyond the NPC and into the nuclear interior.24, 76 Mlp1p is involved in gene targeting as deletion of MLP1 prevents recruitment of GAL10 and HSP104 to the periphery.32 Affinity purification of Mlp1p coupled with mass spectrometry revealed a physical interaction with the Spt-Ada-Gcn5 histone acetyltransferase complex, SAGA.77 Moreover, ChIP experiments revealed that Mlp1p and SAGA components associate at the same promoter region of the GAL genes and that Mlp1p association is dependent on the integrity of the SAGA complex.77 Interestingly, the SAGA component Sus1p is also present in a Sac3p containing mRNA export complex located at the nuclear pore complex and both Sac3p and Sus1p are required for efficient gene recruitment, providing further evidence that an NPC association is an important feature in peripheral gene targeting.29, 73
It appears that multiple aspects of transcriptional activation are required for NPC-chromatin interactions, however NPC-chromatin interactions are not required for all activation events. Therefore, a mechanism must exist that specifies which genes will adopt an NPC association. Such a mechanism is likely to be encoded by _cis_-acting DNA elements as peripheral targeting of some yeast genes are independent of transcription.61, 63 A recent study identified two gene recruitment sequences (GRS I and GRS II) within the promoter region of the INO1 gene responsible for INO1 targeting to the NPC and were sufficient to target an ectopic locus to the nuclear periphery.78 GRS I elements were identified in 94 promoters including several that are recruited to NPCs. The factors that bind GRS elements and promote recruitment remain a mystery but it remains only a matter of time before they too are identified to further inform the spatial organization and regulation of the genome.
Nuclear pore complexes and transcriptional memory
In yeast a number of inducible genes are recruited to nuclear pores following induction. Interestingly, they appear to be retained at the periphery over multiple generations even after transcriptional repression.63 Retention at the nuclear periphery during periods of repression is thought to serve as a memory of previous transcriptional activity to promote an accelerated transcriptional response following reactivation.79 The fact that memory is maintained through multiple cell cycles indicates that the molecular mechanisms responsible are stable through DNA replication and mitosis and ultimately inherited by daughter cells. How do cells remember a previous transcriptional state? Epigenetic changes in chromatin structure are likely responsible. Alterations in nucleosome positioning mediated by the chromatin remodeling complex SWI/SNF and incorporation of the histone variant H2A.Z into assembled nucleosomes both contribute to transcriptional memory.63, 80 An additional mechanism was identified by two recent studies that revealed a link between gene looping and transcriptional memory.81, 82 Gene loops are dynamic structures formed upon transcriptional initiation through interactions between the promoter and 3′-end of a gene. Gene looping has been observed for both yeast and mammalian genes, however not all genes form loops.81, 83, 84 Transcription-dependent gene loop structures were identified in two genes (HXK1 and GAL1) that associate with NPCs during transcriptional activation and gene loops were maintained during intervening periods of transcriptional repression.81 These DNA loop structures are termed memory gene loops (MGLs) and are required for transcriptional memory; mutations that prevent memory gene loop formation permit initial GAL gene activation, but transcriptional memory is lost. The molecular mechanisms that facilitate inheritance of MGLs to daughter cells during mitosis remain unclear. Strategies that establish transcriptional memory are likely to involve changes in chromatin structure through post-translational histone modifications and/or incorporation of histone variants that establish epigenetic marks for MGLs to reform in daughter cells upon completion of mitosis. How histone modifications can establish gene loops and maintain them to ensure their propagation through replication, mitosis and subsequent generations to act as memory markers remain key questions.
A role for the NPC in transcriptional memory is implicated by the association of memory gene loops with the nuclear basket protein Mlp1p.81 Mlp1p associates with memory gene loops through interactions with both the 5′ UAS and 3′-end regions, but not with internal regions of the HXK1 gene. Mlp1p association with memory gene loops is transcription dependent and is maintained for the duration of the transcriptional memory. Furthermore, deletion of MLP1 disrupted memory gene loop formation and resulted in loss of transcriptional memory.81 Gene recruitment to the NPC could enhance memory gene loop formation by acting as a scaffold to promote interactions with both promoter elements and the 3′-end regions of activated genes. Alternatively, the NPC may provide an environment that is conducive for the chromatin reorganization necessary for transcriptional memory as suggested by the physical interaction of Nup2p and H2A.Z57, both of which have been implicated in transcriptional memory of INO1.57, 63
Nuclear pore complexes are facilitators of chromatin boundaries
Chromatin boundary elements are stably maintained and inheritable epigenetic regions of specialized chromatin structure that protect adjacent genes from repressive or activating effects of nearby heterochromatin or enhancer elements, respectively (reviewed in85-87). Although much progress has been made concerning the role of boundary elements, little is known about their composition or mechanism of action. Surprisingly, components of the nuclear pore basket and nuclear transport machinery were identified in a yeast genetic screen as factors exhibiting robust boundary activity by preventing the spread of heterochromatin into a neighboring active region.88 Moreover, boundary activity was dependent on physical interaction with the nuclear pore basket, suggesting that the NPC generates a boundary between repressive and active domains.
Nup2p transcends the classical division between the mobile and stationary phases of the transport machinery by cycling on and off the basket of the NPC in a Ran-dependent manner.74, 75 Deletion of NUP2 abolishes boundary activity of all NPC associated components tested concomitant with loss of an NPC localized reporter gene.57, 88 Interestingly, Nup60p, which is responsible for anchoring Nup2p to the NPC, is also required for Nup2p-dependent boundary activity, suggesting that boundary activity of Nup2p requires attachment to NPCs.57 Proteomic, transcriptomic and genetic studies revealed Nup2p and associated Ran-GEF, Prp20p, physically interact with the boundary proteins histone variant H2A.Z and the chromatin remodeling complex ISW2, and co-isolating chromatin contains epigenetic marks characteristic of boundaries.57 While a separate proteomic study to identify chromatin complexes at boundary regions failed to detect Prp20p or Nup2p, it did identify multiple Prp20p and Nup2p associated proteins.89 Given these results a dynamic model of NPC-mediated boundary activity was proposed.57 In this model the NPC functions as a nexus whereby intranuclear Nup2p associates with chromatin-bound Prp20p at boundary regions, re-association of Nup2p to the nuclear pore basket promotes localization of chromatin to the periphery where the NPC facilitates relocation of chromatin to distinct nuclear subcompartments to stabilize either an active or inactive transcriptional state.
Disease states associated with altered nuclear envelope function
Laminopathies
While studies implicating the three-dimensional architecture of the nucleus have significant importance with respect to understanding gene expression, our understanding of these mechanisms promises to have major implications for human health. For example, the nuclear lamina has long been linked to a class of diseases called laminopathies. Laminopathies are a diverse set of heterozygous genetic disorders that include several types of muscular dystrophies, lipodystrophies and premature ageing syndromes and are caused by alterations of the nuclear lamina (e.g. progeria; see Table 1). A single nucleotide substitution, C-to-T at position 1824 of LMNA prevents removal of a post-translational lipid modification resulting in a permanently farnesylated form of lamin A, termed progerin, which is irreversibly anchored to the INM.90 Progerin acts as a dominant negative mutation by disrupting proper lamin polymerization leading to gross nuclear envelope abnormalities and premature ageing as seen in Hutchinson-Gilford progeria syndrome (HGPS; discussed in Box 4).16 In patient fibroblasts, loss of peripheral heterochromatin is accompanied by epigenetic changes consistent with reduced heterochromatin, specifically a decrease in H3 K9me3, a reduction in co-localization of the heterochromatin markers H3 K9me3 and heterochromatin protein 1 (HP1), and increased expression of a pericentric heterochromatic region.91 These results suggest expression of mutant lamins alter the epigenetic organization of pericentric chromatin resulting in changes in gene expression that are likely to have profound impacts on disease progression.
TABLE 1.
Laminopathies
| Disease | GENE (protein) affected | Clinical relevance | Refs |
|---|---|---|---|
| Hutchinson-Gilford progeria syndrome | LMNA (lamin A) | Premature ageing, impaired growth, alopecia, loss of subcutaneous body fat, atherosclerosis | 90 |
| Emery-Dreifuss muscular dystrophy | LMNA EMD (emerin) | Progressive loss of skeletal muscle, cardiomyopathy, muscle contractures | 138, 139 |
| Charot-Marie-Tooth disease | LMNA | Motor defects in lower limbs, foot deformities and loss of tendon reflexes in lower limbs | 140 |
| Atypical Werner syndrome | LMNA | Premature ageing apparent in early adults, cataracts, atherosclerosis | 141 |
| Limb girdle muscular dystrophy | LMNA | Progressive weakening of shoulder and pelvic muscles, muscle contractures, cardiac arrhythmia | 142 |
| Dilated cardiomyopathy | LMNA | Ventricular dilation, cardiac arrhythmia | 143 |
| Dunnigan-type familial partial lipodystrophy | LMNA | Loss of adipose tissue, insulin-resistant diabetes, atherosclerosis | 144 |
| Mandibuloacral dysplasia | LMNA | Mandibular hypoplasia, delayed cranial development, dental crowding, impaired growth, loss of adipose tissue | 145 |
| Restricted dermopathy | LMNA, ZMPSTE24 | Microstomia, pulmonary hypoplasia | 146, 147 |
| Barraquer-Simons syndrome | LMNB2 (lamin B2) | Loss of adipose tissue, more frequent in females (4:1) | 148 |
| Autosomal dominant leukodystrophy | LMNB2 | Progressive demyelination of central nervous system leading to cerebellar dysfunction | 149 |
| Greenberg dysplasia | LBR (lamin B receptor) | Severe skeletal abnormalities, polydactyly, leads to prenatal death | 150 |
| Pelger-Huet anomaly | LBR | Heterozygous mutation results in benign hypolobulation of granulocyte nuclei; homozygous mutations lead to skeletal abnormalities | 151, 152 |
BOX 4. Hutchinson-Gilford progeria syndrome (HGPS).
Slowed physical growth and rapid aging is the characteristic symptom of patients suffering from Hutchinson-Gilford progeria syndrome (HGPS), an extremely rare disease affecting 1 in 4 million live births, typically identified at an early age (∼12 months). HGPS patients display the following clinical phenotypes: stunted stature, facial-cranial abnormalities, loss of joint mobility, scleroderma, alopecia, loss of subcutaneous body fat (lipodystrophy) but display normal cognitive development. HGPS is fatal with an average life span of 12 years and death attributable to progressive atherosclerosis leading to myocardial infarction or cerebrovascular event. A heterozygous C-to-T substitution at nucleotide 1824 of the lamin A encoding gene LMNA, is associated with 90% of all HGPS patients and although it does not affect the amino acid sequence it introduces a cryptic splice site leading to an mRNA lacking 150 nucleotides.90 The resulting mutant protein, termed progerin, lacks 50 amino acid residues. Removal of the C-terminus also eliminates a cleavage site for the protease ZMPSTE24, preventing this protease from releasing a farnesylated peptide from progerin. Permanent farnesylation of progerin stabilizes its interaction with the NE and is thought to disrupt normal lamin polymerization and function, and potentially manifestation of HGPS.
Whereas alterations in lamina structure clearly impact global gene expression through disruption of lamin-chromatin contacts, gene-specific effects also occur through misregulation of lamina associated transcriptional regulators. Studies of the c-Fos and Oct-1 transcription factors (TFs) have demonstrated that sequestration by the lamina prevents these proteins from binding to downstream target genes, providing both an additional method of negative regulation and a mechanism for rapid gene activation.92-94 Such regulatory mechanisms are likely not limited to c-Fos and Oct-1 as numerous TFs associate with the nuclear lamina including sterol response element binding protein 1 (SREBP1) and retinoblastoma transcriptional regulator (pRb) among others.95-98 In the case of pRb, interactions with lamin A and lamin C facilitate its peripheral localization.95, 97 Mutations that alter lamina structure are likely to affect regulation of associated transcription factors leading to changes in expression of target genes. One mechanism by which pRb regulates gene expression is through recruitment of histone methyltransferases, accordingly pRb is required for H3 K9 methylation.99, 100 This observation suggests that misregulation of pRb may contribute to the epigenetic changes in HGPS cells discussed above. Intriguingly, binding of the transcription factor pRb to the lamina is thought to prevent pRb degradation and promote mesenchymal differentiation of skeletal muscle cells, a tissue often affected by laminopathies.101
Like pRb, interactions between SREBP1 and the lamina are important in regulated cellular differentiation. SREBP1 is a transcription factor required for adipocyte differentiation and overexpression of lamin A impairs this process, whereas differentiation occurs more frequently in lamin A/C deficient mouse embryonic fibroblasts (MEFs).14, 102 Moreover, lamin mutations associated with forms of lipodystrophy displayed reduced binding to SREBP1.98 Misregulation of transcription factors by structural alterations in the nuclear lamina are likely important factors contributing to disease progression in muscular dystrophies and lipidystrophies.101, 103
Nucleoporins and cancer
Mutations in four nucleoporins, Nup88, Nup214, Tpr and Nup98, have been associated with cancers of various types including carcinomas, sarcomas and lymphomas (reviewed in104-107). The mechanisms underlying these phenotypes are unclear; however, the molecular consequences are similar - genes required for proliferation are up-regulated and genes required for differentiation are down-regulated.
Overexpression of NUP88 has been reported in multiple tumors including, but not limited to, carcinomas, sarcomas and lymphomas.108, 109 Nup88 staining of biopsies has been proposed as both a marker of tumor progression and as an indicator of patient prognosis. NUP88 expression increases during tumorigenesis and high expression levels are associated with poorly differentiated tumors and increased aggressiveness of breast and colorectal carcinomas.110, 111 Strikingly, NUP88 overexpression was detected in 76% of ovarian tumors.112 How might overexpression of NUP88 lead to cancer? The mechanism is unclear, however there is evidence that Nup88 is required for import of the transcription factor NF-κB and overexpression may alter nuclear trafficking of NF-κB leading to a nuclear accumulation and constitutive NF-κB activity resulting in up-regulation of target genes involved in cell proliferation.113-115
Nup214, Tpr and Nup98 are associated with multiple forms of leukemia; chromosome translocation events involving these genes give rise to oncogenic fusion proteins. Fusion of the nucleoporin Tpr with the receptor tyrosine kinase MET creates a cytoplasmic chimeric protein capable of dimerization via the Tpr domain causing receptor kinase activation and subsequent signaling to downstream MAPK and PI3K pathways.116-118 Loss of negative regulatory elements within the receptor portion of the Tpr-MET fusion permits constitutive signaling generating a transcriptional response promoting cell proliferation (reviewed in119). An alternate mechanism leading to receptor dimerization occurs in 6% of patients suffering from T-cell acute lymphoblastic leukemia through fusion of Nup214 and ABL1, encoding the Abl non-receptor tyrosine kinase.120, 121 Nup214-Abl retains its ability to associate with the cytoplasmic face of the NPC and oncogenic transformation is dependent on interaction with other nucleoporins. Nup214-Abl is unable to self oligomerize; however, the close proximity of multiple chimeric proteins at the NPC is thought to promote constitutive kinase activation inducing cell proliferative pathways including ERK activation and subsequent transcription of the interleukin-2 gene, encoding a mitogen required for T-cell proliferation.120-122 Although the mechanisms that permit dimerization differ between Tpr-MET and Nup214-Abl, both fusions result in constitutive kinase activation and contribute to tumor progression.
A direct role for NPC components in gene regulation of tumorigenesis was revealed in studies of Nup98. Numerous Nup98 oncogenic fusions have been described (Table 2), each consisting of a partner gene encoding either a DNA or chromatin binding protein and the 5′ region of the NUP98 gene, including NUP98 promoter elements and an amino acid sequence rich in phenylalanine-glycine (FG) repeats that recruit the histone acetyltransferases CREB binding protein (CBP) and p300 to promote transcription.123 It is not surprising then, that potent oncogenic Nup98 fusions combine the ability of Nup98's FG region to bind CBP/p300 and transcriptional activators capable of binding DNA, particularly members of the homeodomain family of proteins encoded by the homeobox (HOX) genes involved in regulating cell proliferation and differentiation.
TABLE 2.
Nucleoporin mutations associated with cancer
| Nucleoporin | Fusion partner gene | Partner protein function | refs |
|---|---|---|---|
| NUP88 | N/A | NUP88 overexpression | 108, 109 |
| NUP98 | HOXA9/11/13 | Homeobox transcription factors | 124, 153, 154 |
| HOXC11/13 | Homeobox transcription factors | 155, 156 | |
| HOXD11/13 | Homeobox transcription factors | 157, 158 | |
| NSD1 | Lysine methytransferase | 159 | |
| NSD3 | Lysine methytransferase | 160 | |
| JARID1A | Lysine demethylase | 161 | |
| PRRX1/2 | Homeobox transcription factors | 162, 163 | |
| TOP1 | Topoisomerase 1 | 164 | |
| TOP2B | Topoisomerase 2 | 165 | |
| HHEX | Homeobox transcription factor | 166 | |
| SETBP1 | SET binding protein 1 | 167 | |
| RAP1GDS1 | Rap1 guanine nucleotide exchange factor | 168 | |
| ANKRD28 | Ankyrin repeat domain protein 28 | 169 | |
| NUP214 | DEK | DNA binding phosphoprotein | 170, 171 |
| SET | Histone chaperone | 172 | |
| ABL1 | Non-receptor tyrosine kinase | 120 | |
| TPR | MET | Hepatocyte growth factor receptor | 116, 173 |
| NTRK1 | Nerve growth factor receptor tyrosine kinase | 174 |
In some patients suffering from acute myeloid leukemia (AML) a rare but recurrent translocation event fuses the NUP98 and HOXA9 genes.124, 125 Microarray studies on hematopoietic progenitor cells expressing the NUP98-HOXA9 fusion indicate that important transcriptional targets of the oncogenic fusion protein are HOX genes themselves, and genes functioning in myeloid differentiation, which are up-regulated and down-regulated respectively.126 The importance of HOX genes in nup-mediated cancers are underscored by ChIP studies revealing enrichment of the SET-Nup214 oncogenic fusion protein at HOXA gene promoters in patients suffering from T-cell acute lymphoblastic leukemia.127 Interestingly, although expression of NUP98-HOXA9 in a mouse model of leukemia resulted in increased proliferation and decreased differentiation of hematopoietic progenitor cells, the onset of leukemia took four months longer than expression of HOXA9 alone.128, 129 This result suggests that additional genetic factors are required for full disease progression.
Systems approaches to identify novel inner nuclear membrane proteins
Despite the fact that numerous diseases are attributable to defects in the nuclear envelope, a molecular understanding of the myriad of functions carried out by the nuclear envelope architecture remains somewhat vague. Defects in the nuclear membrane may contribute to disease through increased fragility of the nucleus under mechanical stress, alterations in chromatin structure, or misregulation of transcription factors, all of which culminate in changes in gene expression. It seems likely that any one of these mechanisms could contribute to disease. To gain a better understanding the nuclear envelope and its contributions to normal and altered gene expression patterns will require further characterization of the nuclear envelope proteome, its dynamics and interactions with chromatin. Indeed, proteomics studies have begun to illuminate these issues. With the realization of the human genome project, in silico based computational approaches have identified a small number of new nuclear envelope components. For example, Nesprin-2 was identified as a gene with high similarity to the human ONM protein Nesprin-1130, whereas the mammalian proteins Sun1 and Sun2 were found to be highly homologous to the INM protein Unc84 of Caenorhabditis elegans.131 Interactions between members of the actin-binding Nesprin and lamin-binding Sun family proteins provide a physical connection between the cytoskeleton and nuclear lamina. Importantly, mutations associated with laminopathies disrupt these connections.132, 133
Mass spectrometric methods also hold promise for further mechanistic insights. As in any biochemical organelle characterization, a major challenge is to distinguish bona fide components of the organelle from contaminants. The nuclear envelope presents a unique complication - the cytoplasmic sheet of the NE is contiguous with the ER. To overcome this problem Schirmer and colleagues134 used a subtractive hi-throughput shotgun proteomics approach in which nuclear envelope and ER fractions enriched for integral membrane proteins were obtained from rat livers and protein compositions were independently analyzed by multidimensional protein identification technology (MudPIT), which couples tandem mass spectrometry with liquid chromatography to allow analysis of complex biochemical fractions.135, 136 Proteins identified in both fractions were subtracted from the nuclear envelope fraction, leaving the remaining proteins as possible constituents of the inner nuclear membrane. Using this approach the authors identified 67 novel putative inner nuclear membrane proteins encoding predicted transmembrane domains. Further microscopy analysis confirmed that 8 of 8 candidates tested were localized to the nuclear envelope. Importantly, only 5 of these novel inner membrane proteins did not have an apparent mammalian homolog and, interestingly, 23 genes mapped to chromosome regions linked to a variety of dystrophies with similarity to laminopathies. There is little doubt that molecular characterization of these proteins will lead to a better understanding of the molecular bases of laminopathies and how the 3D architecture of the nucleus and the physical constraints of the NE impact complex control of the genome.
Future perspectives
It is clear that to understand the complexities of gene regulation we cannot simply view chromatin as a platform onto which regulatory factors bind and release. Indeed, chromatin is highly dynamic and its movement within the nucleus plays a major role in defining, maintaining and switching its activity states. Accordingly, the nuclear architecture itself imparts control on the genome. While we now appreciate these influences, the rules of the game remain elusive. The nuclear periphery does not simply appear to define a state, such as “ON or OFF”; it seems to form part of an organizational domain structure comprised of regions that both promote and prevent gene expression. In addition, studies in yeast reveal that the nuclear pore complex can function to separate active and repressed genes - acting as boundaries between these domains. Moreover, NPCs play a role in transcriptional memory, ensuring cells remember their expression states so that they can retain expression patterns through division and reproduce expression states in future generations.
Yeast has certainly been a tremendously valuable tool to get a foothold into the cell biology of transcriptional control; however, mammalian nuclei are much greater in volume, house more highly organized (hetero)chromatin, dedicate more activity to post-transcriptional control mechanisms, undergo an open mitosis wherein the nuclear envelope completely breaks down at mitosis, and have a significant nuclear lamina structure lining the inner nuclear membrane. We must therefore ask to what extent lessons from yeast can be extended to humans. For example, we presume that in yeast random movements appear to be sufficient for chromatin to sample large portions of the nuclear volume and that a retention mechanism located at the NE may suffice to restrict movements within the nuclear periphery. But given the large size of mammalian nuclei, do mechanisms exist within the interphase nucleus of mammalian cells that mediate long range gene trafficking? Are there specific (energy dependent) mechanisms at play or does the activity depend on (facilitated) diffusion? In the face of a mitotic dissassembly of the organizational structure of the nuclear periphery, what role(s) do the mammalian nuclear periphery play in transcriptional memory, switching transcriptional states, or differentiation? How specific are the influences of the periphery with respect to the expression of gene classes or during development?
While the mechanisms of the phenomena in which nuclear organization influences gene expression are yet to be elucidated, the expanding list of human diseases attributable to nuclear organization based-misregulation reminds us that the answers to these questions are critical.
Acknowledgments
We thank members of the Aitchison and Wozniak laboratories for helpful comments. This work was supported by grants GM075152, GMO76547, and RR022220 from the U.S. National Institutes of Health to John Aitchison and grants CIHR 12547, CIHR 36519, HHMI International Investigator award to Rick Wozniak.
References
- 1.Heitz E. Das Heterochromatin der Moose. Jahrb Wiss Bot. 1928;69:762–818. [Google Scholar]
- 2.Passarge E. Emil Heitz and the concept of heterochromatin: longitudinal chromosome differentiation was recognized fifty years ago. Am J Hum Genet. 1979;31(2):106–115. [PMC free article] [PubMed] [Google Scholar]
- 3.Cairns BR. The logic of chromatin architecture and remodelling at promoters. Nature. 2009;461(7261):193–198. doi: 10.1038/nature08450. [DOI] [PubMed] [Google Scholar]
- 4.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128(4):707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 5.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 6.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 7.Wan Y, Chiang JH, Lin CH, Arens CE, Saleem RA, Smith JJ, Aitchison JD. Histone chaperone Chz1p regulates H2B ubiquitination and subtelomeric anti-silencing. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkp1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shia WJ, Li B, Workman JL. SAS-mediated acetylation of histone H4 Lys 16 is required for H2A.Z incorporation at subtelomeric regions in Saccharomyces cerevisiae. Genes Dev. 2006;20(18):2507–2512. doi: 10.1101/gad.1439206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Babiarz JE, Halley JE, Rine J. Telomeric heterochromatin boundaries require NuA4-dependent acetylation of histone variant H2A.Z in Saccharomyces cerevisiae. Genes Dev. 2006;20(6):700–710. doi: 10.1101/gad.1386306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blobel G. Gene gating: a hypothesis. Proc Natl Acad Sci U S A. 1985;82(24):8527–8529. doi: 10.1073/pnas.82.24.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Comings DE, Okada TA. Association of chromatin fibers with the annuli of the nuclear membrane. Exp Cell Res. 1970;62(2):293–302. doi: 10.1016/0014-4827(70)90557-4. [DOI] [PubMed] [Google Scholar]
- 12.Andres V, Gonzalez JM. Role of A-type lamins in signaling, transcription, and chromatin organization. J Cell Biol. 2009;187(7):945–957. doi: 10.1083/jcb.200904124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. The nuclear lamina comes of age. Nat Rev Mol Cell Biol. 2005;6(1):21–31. doi: 10.1038/nrm1550. [DOI] [PubMed] [Google Scholar]
- 14.Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22(7):832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herrmann H, Bar H, Kreplak L, Strelkov SV, Aebi U. Intermediate filaments: from cell architecture to nanomechanics. Nat Rev Mol Cell Biol. 2007;8(7):562–573. doi: 10.1038/nrm2197. [DOI] [PubMed] [Google Scholar]
- 16.Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101(24):8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rowat AC, Lammerding J, Herrmann H, Aebi U. Towards an integrated understanding of the structure and mechanics of the cell nucleus. Bioessays. 2008;30(3):226–236. doi: 10.1002/bies.20720. [DOI] [PubMed] [Google Scholar]
- 18.Dauer WT, Worman HJ. The nuclear envelope as a signaling node in development and disease. Dev Cell. 2009;17(5):626–638. doi: 10.1016/j.devcel.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 19.Roux KJ, Burke B. Nuclear envelope defects in muscular dystrophy. Biochim Biophys Acta. 2007;1772(2):118–127. doi: 10.1016/j.bbadis.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet. 2006;7(12):940–952. doi: 10.1038/nrg1906. [DOI] [PubMed] [Google Scholar]
- 21.Dechat T, Adam SA, Goldman RD. Nuclear lamins and chromatin: when structure meets function. Adv Enzyme Regul. 2009;49(1):157–166. doi: 10.1016/j.advenzreg.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Worman HJ, Bonne G. “Laminopathies”: a wide spectrum of human diseases. Exp Cell Res. 2007;313(10):2121–2133. doi: 10.1016/j.yexcr.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, Sali A, Rout MP. The molecular architecture of the nuclear pore complex. Nature. 2007;450(7170):695–701. doi: 10.1038/nature06405. [DOI] [PubMed] [Google Scholar]
- 24.Strambio-de-Castillia C, Blobel G, Rout MP. Proteins connecting the nuclear pore complex with the nuclear interior. J Cell Biol. 1999;144(5):839–855. doi: 10.1083/jcb.144.5.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frosst P, Guan T, Subauste C, Hahn K, Gerace L. Tpr is localized within the nuclear basket of the pore complex and has a role in nuclear protein export. J Cell Biol. 2002;156(4):617–630. doi: 10.1083/jcb.200106046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jhunjhunwala S, van Zelm MC, Peak MM, Murre C. Chromatin architecture and the generation of antigen receptor diversity. Cell. 2009;138(3):435–448. doi: 10.1016/j.cell.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kosak ST, Skok JA, Medina KL, Riblet R, Le Beau MM, Fisher AG, Singh H. Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science. 2002;296(5565):158–162. doi: 10.1126/science.1068768. [DOI] [PubMed] [Google Scholar]
- 28.Taddei A, Van Houwe G, Hediger F, Kalck V, Cubizolles F, Schober H, Gasser SM. Nuclear pore association confers optimal expression levels for an inducible yeast gene. Nature. 2006;441(7094):774–778. doi: 10.1038/nature04845. [DOI] [PubMed] [Google Scholar]
- 29.Cabal GG, Genovesio A, Rodriguez-Navarro S, Zimmer C, Gadal O, Lesne A, Buc H, Feuerbach-Fournier F, Olivo-Marin JC, Hurt EC, Nehrbass U. SAGA interacting factors confine sub-diffusion of transcribed genes to the nuclear envelope. Nature. 2006;441(7094):770–773. doi: 10.1038/nature04752. [DOI] [PubMed] [Google Scholar]
- 30.Brickner JH, Walter P. Gene recruitment of the activated INO1 locus to the nuclear membrane. PLoS Biol. 2004;2(11):e342. doi: 10.1371/journal.pbio.0020342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Casolari JM, Brown CR, Drubin DA, Rando OJ, Silver PA. Developmentally induced changes in transcriptional program alter spatial organization across chromosomes. Genes Dev. 2005;19(10):1188–1198. doi: 10.1101/gad.1307205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dieppois G, Iglesias N, Stutz F. Cotranscriptional recruitment to the mRNA export receptor Mex67p contributes to nuclear pore anchoring of activated genes. Mol Cell Biol. 2006;26(21):7858–7870. doi: 10.1128/MCB.00870-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarma NJ, Haley TM, Barbara KE, Buford TD, Willis KA, Santangelo GM. Glucose-responsive regulators of gene expression in Saccharomyces cerevisiae function at the nuclear periphery via a reverse recruitment mechanism. Genetics. 2007;175(3):1127–1135. doi: 10.1534/genetics.106.068932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abruzzi KC, Belostotsky DA, Chekanova JA, Dower K, Rosbash M. 3′-end formation signals modulate the association of genes with the nuclear periphery as well as mRNP dot formation. EMBO J. 2006;25(18):4253–4262. doi: 10.1038/sj.emboj.7601305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA. Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol. 1999;145(6):1119–1131. doi: 10.1083/jcb.145.6.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bolzer A, Kreth G, Solovei I, Koehler D, Saracoglu K, Fauth C, Muller S, Eils R, Cremer C, Speicher MR, Cremer T. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 2005;3(5):e157. doi: 10.1371/journal.pbio.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reddy KL, Zullo JM, Bertolino E, Singh H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature. 2008;452(7184):243–247. doi: 10.1038/nature06727. [DOI] [PubMed] [Google Scholar]
- 38.Williams RR, Azuara V, Perry P, Sauer S, Dvorkina M, Jorgensen H, Roix J, McQueen P, Misteli T, Merkenschlager M, Fisher AG. Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J Cell Sci. 2006;119(Pt 1):132–140. doi: 10.1242/jcs.02727. [DOI] [PubMed] [Google Scholar]
- 39.Ragoczy T, Bender MA, Telling A, Byron R, Groudine M. The locus control region is required for association of the murine beta-globin locus with engaged transcription factories during erythroid maturation. Genes Dev. 2006;20(11):1447–1457. doi: 10.1101/gad.1419506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiblin AE, Cui W, Clark AJ, Bickmore WA. Distinctive nuclear organisation of centromeres and regions involved in pluripotency in human embryonic stem cells. J Cell Sci. 2005;118(Pt 17):3861–3868. doi: 10.1242/jcs.02500. [DOI] [PubMed] [Google Scholar]
- 41.Jin Q, Trelles-Sticken E, Scherthan H, Loidl J. Yeast nuclei display prominent centromere clustering that is reduced in nondividing cells and in meiotic prophase. J Cell Biol. 1998;141(1):21–29. doi: 10.1083/jcb.141.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Funabiki H, Hagan I, Uzawa S, Yanagida M. Cell cycle-dependent specific positioning and clustering of centromeres and telomeres in fission yeast. J Cell Biol. 1993;121(5):961–976. doi: 10.1083/jcb.121.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akhtar A, Gasser SM. The nuclear envelope and transcriptional control. Nat Rev Genet. 2007;8(7):507–517. doi: 10.1038/nrg2122. [DOI] [PubMed] [Google Scholar]
- 44.Gotta M, Laroche T, Formenton A, Maillet L, Scherthan H, Gasser SM. The clustering of telomeres and colocalization with Rap1, Sir3, and Sir4 proteins in wild-type Saccharomyces cerevisiae. J Cell Biol. 1996;134(6):1349–1363. doi: 10.1083/jcb.134.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maillet L, Boscheron C, Gotta M, Marcand S, Gilson E, Gasser SM. Evidence for silencing compartments within the yeast nucleus: a role for telomere proximity and Sir protein concentration in silencer-mediated repression. Genes Dev. 1996;10(14):1796–1811. doi: 10.1101/gad.10.14.1796. [DOI] [PubMed] [Google Scholar]
- 46.Palladino F, Laroche T, Gilson E, Axelrod A, Pillus L, Gasser SM. SIR3 and SIR4 proteins are required for the positioning and integrity of yeast telomeres. Cell. 1993;75(3):543–555. doi: 10.1016/0092-8674(93)90388-7. [DOI] [PubMed] [Google Scholar]
- 47.Towbin BD, Meister P, Gasser SM. The nuclear envelope--a scaffold for silencing? Curr Opin Genet Dev. 2009;19(2):180–186. doi: 10.1016/j.gde.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 48.Buhler M, Gasser SM. Silent chromatin at the middle and ends: lessons from yeasts. EMBO J. 2009;28(15):2149–2161. doi: 10.1038/emboj.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taddei A, Gasser SM. Multiple pathways for telomere tethering: functional implications of subnuclear position for heterochromatin formation. Biochim Biophys Acta. 2004;1677(1-3):120–128. doi: 10.1016/j.bbaexp.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 50.Taddei A, Van Houwe G, Nagai S, Erb I, van Nimwegen E, Gasser SM. The functional importance of telomere clustering: global changes in gene expression result from SIR factor dispersion. Genome Res. 2009;19(4):611–625. doi: 10.1101/gr.083881.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andrulis ED, Neiman AM, Zappulla DC, Sternglanz R. Perinuclear localization of chromatin facilitates transcriptional silencing. Nature. 1998;394(6693):592–595. doi: 10.1038/29100. [DOI] [PubMed] [Google Scholar]
- 52.Luderus ME, de Graaf A, Mattia E, den Blaauwen JL, Grande MA, de Jong L, van Driel R. Binding of matrix attachment regions to lamin B1. Cell. 1992;70(6):949–959. doi: 10.1016/0092-8674(92)90245-8. [DOI] [PubMed] [Google Scholar]
- 53.Zhao K, Harel A, Stuurman N, Guedalia D, Gruenbaum Y. Binding of matrix attachment regions to nuclear lamin is mediated by the rod domain and depends on the lamin polymerization state. FEBS Lett. 1996;380(1-2):161–164. doi: 10.1016/0014-5793(96)00034-8. [DOI] [PubMed] [Google Scholar]
- 54.Pickersgill H, Kalverda B, de Wit E, Talhout W, Fornerod M, van Steensel B. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet. 2006;38(9):1005–1014. doi: 10.1038/ng1852. [DOI] [PubMed] [Google Scholar]
- 55.Somech R, Shaklai S, Geller O, Amariglio N, Simon AJ, Rechavi G, Gal-Yam EN. The nuclear-envelope protein and transcriptional repressor LAP2beta interacts with HDAC3 at the nuclear periphery, and induces histone H4 deacetylation. J Cell Sci. 2005;118(Pt 17):4017–4025. doi: 10.1242/jcs.02521. [DOI] [PubMed] [Google Scholar]
- 56.Casolari JM, Brown CR, Komili S, West J, Hieronymus H, Silver PA. Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell. 2004;117(4):427–439. doi: 10.1016/s0092-8674(04)00448-9. [DOI] [PubMed] [Google Scholar]
- 57.Dilworth DJ, Tackett AJ, Rogers RS, Yi EC, Christmas RH, Smith JJ, Siegel AF, Chait BT, Wozniak RW, Aitchison JD. The mobile nucleoporin Nup2p and chromatin-bound Prp20p function in endogenous NPC-mediated transcriptional control. J Cell Biol. 2005;171(6):955–965. doi: 10.1083/jcb.200509061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee A, Tam R, Belhumeur P, DiPaolo T, Clark MW. Prp20, the Saccharomyces cerevisiae homolog of the regulator of chromosome condensation, RCC1, interacts with double-stranded DNA through a multi-component complex containing GTP-binding proteins. J Cell Sci. 1993;106(Pt 1):287–298. doi: 10.1242/jcs.106.1.287. [DOI] [PubMed] [Google Scholar]
- 59.Aebi M, Clark MW, Vijayraghavan U, Abelson J. A yeast mutant, PRP20, altered in mRNA metabolism and maintenance of the nuclear structure, is defective in a gene homologous to the human gene RCC1 which is involved in the control of chromosome condensation. Mol Gen Genet. 1990;224(1):72–80. doi: 10.1007/BF00259453. [DOI] [PubMed] [Google Scholar]
- 60.Berger AB, Cabal GG, Fabre E, Duong T, Buc H, Nehrbass U, Olivo-Marin JC, Gadal O, Zimmer C. High-resolution statistical mapping reveals gene territories in live yeast. Nat Methods. 2008;5(12):1031–1037. doi: 10.1038/nmeth.1266. [DOI] [PubMed] [Google Scholar]
- 61.Schmid M, Arib G, Laemmli C, Nishikawa J, Durussel T, Laemmli UK. Nup-PI: the nucleopore-promoter interaction of genes in yeast. Mol Cell. 2006;21(3):379–391. doi: 10.1016/j.molcel.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 62.Taddei A. Active genes at the nuclear pore complex. Curr Opin Cell Biol. 2007;19(3):305–310. doi: 10.1016/j.ceb.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 63.Brickner DG, Cajigas I, Fondufe-Mittendorf Y, Ahmed S, Lee PC, Widom J, Brickner JH. H2A.Z-mediated localization of genes at the nuclear periphery confers epigenetic memory of previous transcriptional state. PLoS Biol. 2007;5(4):e81. doi: 10.1371/journal.pbio.0050081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brown CR, Kennedy CJ, Delmar VA, Forbes DJ, Silver PA. Global histone acetylation induces functional genomic reorganization at mammalian nuclear pore complexes. Genes Dev. 2008;22(5):627–639. doi: 10.1101/gad.1632708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mendjan S, Taipale M, Kind J, Holz H, Gebhardt P, Schelder M, Vermeulen M, Buscaino A, Duncan K, Mueller J, Wilm M, Stunnenberg HG, Saumweber H, Akhtar A. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol Cell. 2006;21(6):811–823. doi: 10.1016/j.molcel.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 66.Vaquerizas JM, Suyama R, Kind J, Miura K, Luscombe NM, Akhtar A. Nuclear pore proteins nup153 and megator define transcriptionally active regions in the Drosophila genome. PLoS Genet. 2010;6(2):e1000846. doi: 10.1371/journal.pgen.1000846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kurshakova MM, Krasnov AN, Kopytova DV, Shidlovskii YV, Nikolenko JV, Nabirochkina EN, Spehner D, Schultz P, Tora L, Georgieva SG. SAGA and a novel Drosophila export complex anchor efficient transcription and mRNA export to NPC. EMBO J. 2007;26(24):4956–4965. doi: 10.1038/sj.emboj.7601901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Capelson M, Liang Y, Schulte R, Mair W, Wagner U, Hetzer MW. Chromatin-Bound Nuclear Pore Components Regulate Gene Expression in Higher Eukaryotes. Cell. 2010;140(3):372–383. doi: 10.1016/j.cell.2009.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kalverda B, Pickersgill H, Shloma VV, Fornerod M. Nucleoporins Directly Stimulate Expression of Developmental and Cell-Cycle Genes Inside the Nucleoplasm. Cell. 2010;140(3):360–371. doi: 10.1016/j.cell.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 70.Menon BB, Sarma NJ, Pasula S, Deminoff SJ, Willis KA, Barbara KE, Andrews B, Santangelo GM. Reverse recruitment: the Nup84 nuclear pore subcomplex mediates Rap1/Gcr1/Gcr2 transcriptional activation. Proc Natl Acad Sci U S A. 2005;102(16):5749–5754. doi: 10.1073/pnas.0501768102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Drubin DA, Garakani AM, Silver PA. Motion as a phenotype: the use of live-cell imaging and machine visual screening to characterize transcription-dependent chromosome dynamics. BMC Cell Biol. 2006;7:19. doi: 10.1186/1471-2121-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fischer T, Strasser K, Racz A, Rodriguez-Navarro S, Oppizzi M, Ihrig P, Lechner J, Hurt E. The mRNA export machinery requires the novel Sac3p-Thp1p complex to dock at the nucleoplasmic entrance of the nuclear pores. EMBO J. 2002;21(21):5843–5852. doi: 10.1093/emboj/cdf590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rodriguez-Navarro S, Fischer T, Luo MJ, Antunez O, Brettschneider S, Lechner J, Perez-Ortin JE, Reed R, Hurt E. Sus1, a functional component of the SAGA histone acetylase complex and the nuclear pore-associated mRNA export machinery. Cell. 2004;116(1):75–86. doi: 10.1016/s0092-8674(03)01025-0. [DOI] [PubMed] [Google Scholar]
- 74.Dilworth DJ, Suprapto A, Padovan JC, Chait BT, Wozniak RW, Rout MP, Aitchison JD. Nup2p dynamically associates with the distal regions of the yeast nuclear pore complex. J Cell Biol. 2001;153(7):1465–1478. doi: 10.1083/jcb.153.7.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Denning D, Mykytka B, Allen NP, Huang L, Al B, Rexach M. The nucleoporin Nup60p functions as a Gsp1p-GTP-sensitive tether for Nup2p at the nuclear pore complex. J Cell Biol. 2001;154(5):937–950. doi: 10.1083/jcb.200101007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Feuerbach F, Galy V, Trelles-Sticken E, Fromont-Racine M, Jacquier A, Gilson E, Olivo-Marin JC, Scherthan H, Nehrbass U. Nuclear architecture and spatial positioning help establish transcriptional states of telomeres in yeast. Nat Cell Biol. 2002;4(3):214–221. doi: 10.1038/ncb756. [DOI] [PubMed] [Google Scholar]
- 77.Luthra R, Kerr SC, Harreman MT, Apponi LH, Fasken MB, Ramineni S, Chaurasia S, Valentini SR, Corbett AH. Actively transcribed GAL genes can be physically linked to the nuclear pore by the SAGA chromatin modifying complex. J Biol Chem. 2007;282(5):3042–3049. doi: 10.1074/jbc.M608741200. [DOI] [PubMed] [Google Scholar]
- 78.Ahmed S, Brickner DG, Light WH, Cajigas I, McDonough M, Froyshteter AB, Volpe T, Brickner JH. DNA zip codes control an ancient mechanism for gene targeting to the nuclear periphery. Nat Cell Biol. 2010;12(2):111–118. doi: 10.1038/ncb2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brickner JH. Transcriptional memory at the nuclear periphery. Curr Opin Cell Biol. 2009;21(1):127–133. doi: 10.1016/j.ceb.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kundu S, Horn PJ, Peterson CL. SWI/SNF is required for transcriptional memory at the yeast GAL gene cluster. Genes Dev. 2007;21(8):997–1004. doi: 10.1101/gad.1506607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tan-Wong SM, Wijayatilake HD, Proudfoot NJ. Gene loops function to maintain transcriptional memory through interaction with the nuclear pore complex. Genes Dev. 2009;23(22):2610–2624. doi: 10.1101/gad.1823209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laine JP, Singh BN, Krishnamurthy S, Hampsey M. A physiological role for gene loops in yeast. Genes Dev. 2009;23(22):2604–2609. doi: 10.1101/gad.1823609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O'Sullivan JM, Tan-Wong SM, Morillon A, Lee B, Coles J, Mellor J, Proudfoot NJ. Gene loops juxtapose promoters and terminators in yeast. Nat Genet. 2004;36(9):1014–1018. doi: 10.1038/ng1411. [DOI] [PubMed] [Google Scholar]
- 84.Tan-Wong SM, French JD, Proudfoot NJ, Brown MA. Dynamic interactions between the promoter and terminator regions of the mammalian BRCA1 gene. Proc Natl Acad Sci U S A. 2008;105(13):5160–5165. doi: 10.1073/pnas.0801048105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gerasimova TI, Corces VG. Chromatin insulators and boundaries: effects on transcription and nuclear organization. Annu Rev Genet. 2001;35:193–208. doi: 10.1146/annurev.genet.35.102401.090349. [DOI] [PubMed] [Google Scholar]
- 86.West AG, Gaszner M, Felsenfeld G. Insulators: many functions, many mechanisms. Genes Dev. 2002;16(3):271–288. doi: 10.1101/gad.954702. [DOI] [PubMed] [Google Scholar]
- 87.Lunyak VV. Boundaries. Boundaries…Boundaries??? Curr Opin Cell Biol. 2008;20(3):281–287. doi: 10.1016/j.ceb.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 88.Ishii K, Arib G, Lin C, Van Houwe G, Laemmli UK. Chromatin boundaries in budding yeast: the nuclear pore connection. Cell. 2002;109(5):551–562. doi: 10.1016/s0092-8674(02)00756-0. [DOI] [PubMed] [Google Scholar]
- 89.Tackett AJ, Dilworth DJ, Davey MJ, O'Donnell M, Aitchison JD, Rout MP, Chait BT. Proteomic and genomic characterization of chromatin complexes at a boundary. J Cell Biol. 2005;169(1):35–47. doi: 10.1083/jcb.200502104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423(6937):293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, Jenuwein T, Goldman RD. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103(23):8703–8708. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ivorra C, Kubicek M, Gonzalez JM, Sanz-Gonzalez SM, Alvarez-Barrientos A, O'Connor JE, Burke B, Andres V. A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev. 2006;20(3):307–320. doi: 10.1101/gad.349506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gonzalez JM, Navarro-Puche A, Casar B, Crespo P, Andres V. Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. J Cell Biol. 2008;183(4):653–666. doi: 10.1083/jcb.200805049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Malhas AN, Lee CF, Vaux DJ. Lamin B1 controls oxidative stress responses via Oct-1. J Cell Biol. 2009;184(1):45–55. doi: 10.1083/jcb.200804155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mancini MA, Shan B, Nickerson JA, Penman S, Lee WH. The retinoblastoma gene product is a cell cycle-dependent, nuclear matrix-associated protein. Proc Natl Acad Sci U S A. 1994;91(1):418–422. doi: 10.1073/pnas.91.1.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Heessen S, Fornerod M. The inner nuclear envelope as a transcription factor resting place. EMBO Rep. 2007;8(10):914–919. doi: 10.1038/sj.embor.7401075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ozaki T, Saijo M, Murakami K, Enomoto H, Taya Y, Sakiyama S. Complex formation between lamin A and the retinoblastoma gene product: identification of the domain on lamin A required for its interaction. Oncogene. 1994;9(9):2649–2653. [PubMed] [Google Scholar]
- 98.Lloyd DJ, Trembath RC, Shackleton S. A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Hum Mol Genet. 2002;11(7):769–777. doi: 10.1093/hmg/11.7.769. [DOI] [PubMed] [Google Scholar]
- 99.Gonzalo S, Garcia-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, Eguia R, Dean DC, Esteller M, Jenuwein T, Blasco MA. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol. 2005;7(4):420–428. doi: 10.1038/ncb1235. [DOI] [PubMed] [Google Scholar]
- 100.Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, Kouzarides T. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412(6846):561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- 101.Johnson BR, Nitta RT, Frock RL, Mounkes L, Barbie DA, Stewart CL, Harlow E, Kennedy BK. A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Natl Acad Sci U S A. 2004;101(26):9677–9682. doi: 10.1073/pnas.0403250101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Boguslavsky RL, Stewart CL, Worman HJ. Nuclear lamin A inhibits adipocyte differentiation: implications for Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2006;15(4):653–663. doi: 10.1093/hmg/ddi480. [DOI] [PubMed] [Google Scholar]
- 103.Hegele RA. Molecular basis of partial lipodystrophy and prospects for therapy. Trends Mol Med. 2001;7(3):121–126. doi: 10.1016/s1471-4914(01)01930-x. [DOI] [PubMed] [Google Scholar]
- 104.Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26(47):6766–6776. doi: 10.1038/sj.onc.1210760. [DOI] [PubMed] [Google Scholar]
- 105.Slape C, Aplan PD. The role of NUP98 gene fusions in hematologic malignancy. Leuk Lymphoma. 2004;45(7):1341–1350. doi: 10.1080/10428190310001659325. [DOI] [PubMed] [Google Scholar]
- 106.Kau TR, Way JC, Silver PA. Nuclear transport and cancer: from mechanism to intervention. Nat Rev Cancer. 2004;4(2):106–117. doi: 10.1038/nrc1274. [DOI] [PubMed] [Google Scholar]
- 107.Xu S, Powers MA. Nuclear pore proteins and cancer. Semin Cell Dev Biol. 2009;20(5):620–630. doi: 10.1016/j.semcdb.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gould VE, Martinez N, Orucevic A, Schneider J, Alonso A. A novel, nuclear pore-associated, widely distributed molecule overexpressed in oncogenesis and development. Am J Pathol. 2000;157(5):1605–1613. doi: 10.1016/S0002-9440(10)64798-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gould VE, Orucevic A, Zentgraf H, Gattuso P, Martinez N, Alonso A. Nup88 (karyoporin) in human malignant neoplasms and dysplasias: correlations of immunostaining of tissue sections, cytologic smears, and immunoblot analysis. Hum Pathol. 2002;33(5):536–544. doi: 10.1053/hupa.2002.124785. [DOI] [PubMed] [Google Scholar]
- 110.Agudo D, Gomez-Esquer F, Martinez-Arribas F, Nunez-Villar MJ, Pollan M, Schneider J. Nup88 mRNA overexpression is associated with high aggressiveness of breast cancer. Int J Cancer. 2004;109(5):717–720. doi: 10.1002/ijc.20034. [DOI] [PubMed] [Google Scholar]
- 111.Zhang ZY, Zhao ZR, Jiang L, Li JC, Gao YM, Cui DS, Wang CJ, Schneider J, Wang MW, Sun XF. Nup88 expression in normal mucosa, adenoma, primary adenocarcinoma and lymph node metastasis in the colorectum. Tumour Biol. 2007;28(2):93–99. doi: 10.1159/000099154. [DOI] [PubMed] [Google Scholar]
- 112.Martinez N, Alonso A, Moragues MD, Ponton J, Schneider J. The nuclear pore complex protein Nup88 is overexpressed in tumor cells. Cancer Res. 1999;59(21):5408–5411. [PubMed] [Google Scholar]
- 113.Roth P, Xylourgidis N, Sabri N, Uv A, Fornerod M, Samakovlis C. The Drosophila nucleoporin DNup88 localizes DNup214 and CRM1 on the nuclear envelope and attenuates NES-mediated nuclear export. J Cell Biol. 2003;163(4):701–706. doi: 10.1083/jcb.200304046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Uv AE, Roth P, Xylourgidis N, Wickberg A, Cantera R, Samakovlis C. members only encodes a Drosophila nucleoporin required for rel protein import and immune response activation. Genes Dev. 2000;14(15):1945–1957. [PMC free article] [PubMed] [Google Scholar]
- 115.Takahashi N, van Kilsdonk JW, Ostendorf B, Smeets R, Bruggeman SW, Alonso A, van de Loo F, Schneider M, van den Berg WB, Swart GW. Tumor marker nucleoporin 88 kDa regulates nucleocytoplasmic transport of NF-kappaB. Biochem Biophys Res Commun. 2008;374(3):424–430. doi: 10.1016/j.bbrc.2008.06.128. [DOI] [PubMed] [Google Scholar]
- 116.Cooper CS, Blair DG, Oskarsson MK, Tainsky MA, Eader LA, Vande Woude GF. Characterization of human transforming genes from chemically transformed, teratocarcinoma, and pancreatic carcinoma cell lines. Cancer Res. 1984;44(1):1–10. [PubMed] [Google Scholar]
- 117.Park M, Dean M, Cooper CS, Schmidt M, O'Brien SJ, Blair DG, Vande Woude GF. Mechanism of met oncogene activation. Cell. 1986;45(6):895–904. doi: 10.1016/0092-8674(86)90564-7. [DOI] [PubMed] [Google Scholar]
- 118.Rodrigues GA, Park M. Dimerization mediated through a leucine zipper activates the oncogenic potential of the met receptor tyrosine kinase. Mol Cell Biol. 1993;13(11):6711–6722. doi: 10.1128/mcb.13.11.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Peschard P, Park M. From Tpr-Met to Met, tumorigenesis and tubes. Oncogene. 2007;26(9):1276–1285. doi: 10.1038/sj.onc.1210201. [DOI] [PubMed] [Google Scholar]
- 120.Graux C, Cools J, Melotte C, Quentmeier H, Ferrando A, Levine R, Vermeesch JR, Stul M, Dutta B, Boeckx N, Bosly A, Heimann P, Uyttebroeck A, Mentens N, Somers R, MacLeod RA, Drexler HG, Look AT, Gilliland DG, Michaux L, Vandenberghe P, Wlodarska I, Marynen P, Hagemeijer A. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat Genet. 2004;36(10):1084–1089. doi: 10.1038/ng1425. [DOI] [PubMed] [Google Scholar]
- 121.De Keersmaecker K, Rocnik JL, Bernad R, Lee BH, Leeman D, Gielen O, Verachtert H, Folens C, Munck S, Marynen P, Fornerod M, Gilliland DG, Cools J. Kinase activation and transformation by NUP214-ABL1 is dependent on the context of the nuclear pore. Mol Cell. 2008;31(1):134–142. doi: 10.1016/j.molcel.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 122.Zipfel PA, Zhang W, Quiroz M, Pendergast AM. Requirement for Abl kinases in T cell receptor signaling. Curr Biol. 2004;14(14):1222–1231. doi: 10.1016/j.cub.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 123.Kasper LH, Brindle PK, Schnabel CA, Pritchard CE, Cleary ML, van Deursen JM. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol. 1999;19(1):764–776. doi: 10.1128/mcb.19.1.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Borrow J, Shearman AM, Stanton VP, Jr, Becher R, Collins T, Williams AJ, Dube I, Katz F, Kwong YL, Morris C, Ohyashiki K, Toyama K, Rowley J, Housman DE. The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat Genet. 1996;12(2):159–167. doi: 10.1038/ng0296-159. [DOI] [PubMed] [Google Scholar]
- 125.Nakamura T, Largaespada DA, Lee MP, Johnson LA, Ohyashiki K, Toyama K, Chen SJ, Willman CL, Chen IM, Feinberg AP, Jenkins NA, Copeland NG, Shaughnessy JD., Jr Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nat Genet. 1996;12(2):154–158. doi: 10.1038/ng0296-154. [DOI] [PubMed] [Google Scholar]
- 126.Takeda A, Goolsby C, Yaseen NR. NUP98-HOXA9 induces long-term proliferation and blocks differentiation of primary human CD34+ hematopoietic cells. Cancer Res. 2006;66(13):6628–6637. doi: 10.1158/0008-5472.CAN-06-0458. [DOI] [PubMed] [Google Scholar]
- 127.Van Vlierberghe P, van Grotel M, Tchinda J, Lee C, Beverloo HB, van der Spek PJ, Stubbs A, Cools J, Nagata K, Fornerod M, Buijs-Gladdines J, Horstmann M, van Wering ER, Soulier J, Pieters R, Meijerink JP. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood. 2008;111(9):4668–4680. doi: 10.1182/blood-2007-09-111872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kroon E, Thorsteinsdottir U, Mayotte N, Nakamura T, Sauvageau G. NUP98-HOXA9 expression in hemopoietic stem cells induces chronic and acute myeloid leukemias in mice. EMBO J. 2001;20(3):350–361. doi: 10.1093/emboj/20.3.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Iwasaki M, Kuwata T, Yamazaki Y, Jenkins NA, Copeland NG, Osato M, Ito Y, Kroon E, Sauvageau G, Nakamura T. Identification of cooperative genes for NUP98-HOXA9 in myeloid leukemogenesis using a mouse model. Blood. 2005;105(2):784–793. doi: 10.1182/blood-2004-04-1508. [DOI] [PubMed] [Google Scholar]
- 130.Apel ED, Lewis RM, Grady RM, Sanes JR. Syne-1, a dystrophin- and Klarsicht-related protein associated with synaptic nuclei at the neuromuscular junction. J Biol Chem. 2000;275(41):31986–31995. doi: 10.1074/jbc.M004775200. [DOI] [PubMed] [Google Scholar]
- 131.Malone CJ, Fixsen WD, Horvitz HR, Han M. UNC-84 localizes to the nuclear envelope and is required for nuclear migration and anchoring during C. elegans development. Development. 1999;126(14):3171–3181. doi: 10.1242/dev.126.14.3171. [DOI] [PubMed] [Google Scholar]
- 132.Haque F, Lloyd DJ, Smallwood DT, Dent CL, Shanahan CM, Fry AM, Trembath RC, Shackleton S. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol. 2006;26(10):3738–3751. doi: 10.1128/MCB.26.10.3738-3751.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Haque F, Mazzeo D, Patel JT, Smallwood DT, Ellis JA, Shanahan CM, Shackleton S. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem. 2010;285(5):3487–3498. doi: 10.1074/jbc.M109.071910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schirmer EC, Florens L, Guan T, Yates JR, 3rd, Gerace L. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science. 2003;301(5638):1380–1382. doi: 10.1126/science.1088176. [DOI] [PubMed] [Google Scholar]
- 135.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19(3):242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 136.Delahunty CM, Yates JR., 3rd MudPIT: multidimensional protein identification technology. Biotechniques. 2007;43(5):563, 565, 567. passim. [PubMed] [Google Scholar]
- 137.Straight AF, Belmont AS, Robinett CC, Murray AW. GFP tagging of budding yeast chromosomes reveals that protein-protein interactions can mediate sister chromatid cohesion. Curr Biol. 1996;6(12):1599–1608. doi: 10.1016/s0960-9822(02)70783-5. [DOI] [PubMed] [Google Scholar]
- 138.Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8(4):323–327. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- 139.Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21(3):285–288. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 140.De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, Szepetowski P, Hammadouche T, Vandenberghe A, Stewart CL, Grid D, Levy N. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70(3):726–736. doi: 10.1086/339274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, Botha EG, Garg A, Hanson NB, Martin GM, Mian IS, Kennedy BK, Oshima J. LMNA mutations in atypical Werner's syndrome. Lancet. 2003;362(9382):440–445. doi: 10.1016/S0140-6736(03)14069-X. [DOI] [PubMed] [Google Scholar]
- 142.Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B) Hum Mol Genet. 2000;9(9):1453–1459. doi: 10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- 143.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341(23):1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 144.Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9(1):109–112. doi: 10.1093/hmg/9.1.109. [DOI] [PubMed] [Google Scholar]
- 145.Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D'Apice MR, Massart C, Capon F, Sbraccia P, Federici M, Lauro R, Tudisco C, Pallotta R, Scarano G, Dallapiccola B, Merlini L, Bonne G. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71(2):426–431. doi: 10.1086/341908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Navarro CL, De Sandre-Giovannoli A, Bernard R, Boccaccio I, Boyer A, Genevieve D, Hadj-Rabia S, Gaudy-Marqueste C, Smitt HS, Vabres P, Faivre L, Verloes A, Van Essen T, Flori E, Hennekam R, Beemer FA, Laurent N, Le Merrer M, Cau P, Levy N. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13(20):2493–2503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- 147.Navarro CL, Cadinanos J, De Sandre-Giovannoli A, Bernard R, Courrier S, Boccaccio I, Boyer A, Kleijer WJ, Wagner A, Giuliano F, Beemer FA, Freije JM, Cau P, Hennekam RC, Lopez-Otin C, Badens C, Levy N. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum Mol Genet. 2005;14(11):1503–1513. doi: 10.1093/hmg/ddi159. [DOI] [PubMed] [Google Scholar]
- 148.Hegele RA, Cao H, Liu DM, Costain GA, Charlton-Menys V, Rodger NW, Durrington PN. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet. 2006;79(2):383–389. doi: 10.1086/505885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38(10):1114–1123. doi: 10.1038/ng1872. [DOI] [PubMed] [Google Scholar]
- 150.Waterham HR, Koster J, Mooyer P, Noort Gv G, Kelley RI, Wilcox WR, Wanders RJ, Hennekam RC, Oosterwijk JC. Autosomal recessive HEM/Greenberg skeletal dysplasia is caused by 3 beta-hydroxysterol delta 14-reductase deficiency due to mutations in the lamin B receptor gene. Am J Hum Genet. 2003;72(4):1013–1017. doi: 10.1086/373938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Oosterwijk JC, Mansour S, van Noort G, Waterham HR, Hall CM, Hennekam RC. Congenital abnormalities reported in Pelger-Huet homozygosity as compared to Greenberg/HEM dysplasia: highly variable expression of allelic phenotypes. J Med Genet. 2003;40(12):937–941. doi: 10.1136/jmg.40.12.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hoffmann K, Dreger CK, Olins AL, Olins DE, Shultz LD, Lucke B, Karl H, Kaps R, Muller D, Vaya A, Aznar J, Ware RE, Sotelo Cruz N, Lindner TH, Herrmann H, Reis A, Sperling K. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huet anomaly) Nat Genet. 2002;31(4):410–414. doi: 10.1038/ng925. [DOI] [PubMed] [Google Scholar]
- 153.Fujino T, Suzuki A, Ito Y, Ohyashiki K, Hatano Y, Miura I, Nakamura T. Single-translocation and double-chimeric transcripts: detection of NUP98-HOXA9 in myeloid leukemias with HOXA11 or HOXA13 breaks of the chromosomal translocation t(7;11)(p15;p15) Blood. 2002;99(4):1428–1433. doi: 10.1182/blood.v99.4.1428. [DOI] [PubMed] [Google Scholar]
- 154.Suzuki A, Ito Y, Sashida G, Honda S, Katagiri T, Fujino T, Nakamura T, Ohyashiki K. t(7;11)(p15;p15) Chronic myeloid leukaemia developed into blastic transformation showing a novel NUP98/HOXA11 fusion. Br J Haematol. 2002;116(1):170–172. doi: 10.1046/j.1365-2141.2002.03246.x. [DOI] [PubMed] [Google Scholar]
- 155.Taketani T, Taki T, Shibuya N, Kikuchi A, Hanada R, Hayashi Y. Novel NUP98-HOXC11 fusion gene resulted from a chromosomal break within exon 1 of HOXC11 in acute myeloid leukemia with t(11;12)(p15;q13) Cancer Res. 2002;62(16):4571–4574. [PubMed] [Google Scholar]
- 156.La Starza R, Trubia M, Crescenzi B, Matteucci C, Negrini M, Martelli MF, Pelicci PG, Mecucci C. Human homeobox gene HOXC13 is the partner of NUP98 in adult acute myeloid leukemia with t(11;12)(p15;q13) Genes Chromosomes Cancer. 2003;36(4):420–423. doi: 10.1002/gcc.10182. [DOI] [PubMed] [Google Scholar]
- 157.Taketani T, Taki T, Shibuya N, Ito E, Kitazawa J, Terui K, Hayashi Y. The HOXD11 gene is fused to the NUP98 gene in acute myeloid leukemia with t(2;11)(q31;p15) Cancer Res. 2002;62(1):33–37. [PubMed] [Google Scholar]
- 158.Raza-Egilmez SZ, Jani-Sait SN, Grossi M, Higgins MJ, Shows TB, Aplan PD. NUP98-HOXD13 gene fusion in therapy-related acute myelogenous leukemia. Cancer Res. 1998;58(19):4269–4273. [PubMed] [Google Scholar]
- 159.Jaju RJ, Fidler C, Haas OA, Strickson AJ, Watkins F, Clark K, Cross NC, Cheng JF, Aplan PD, Kearney L, Boultwood J, Wainscoat JS. A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood. 2001;98(4):1264–1267. doi: 10.1182/blood.v98.4.1264. [DOI] [PubMed] [Google Scholar]
- 160.Rosati R, La Starza R, Veronese A, Aventin A, Schwienbacher C, Vallespi T, Negrini M, Martelli MF, Mecucci C. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15) Blood. 2002;99(10):3857–3860. doi: 10.1182/blood.v99.10.3857. [DOI] [PubMed] [Google Scholar]
- 161.van Zutven LJ, Onen E, Velthuizen SC, van Drunen E, von Bergh AR, van den Heuvel-Eibrink MM, Veronese A, Mecucci C, Negrini M, de Greef GE, Beverloo HB. Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes Chromosomes Cancer. 2006;45(5):437–446. doi: 10.1002/gcc.20308. [DOI] [PubMed] [Google Scholar]
- 162.Nakamura T, Yamazaki Y, Hatano Y, Miura I. NUP98 is fused to PMX1 homeobox gene in human acute myelogenous leukemia with chromosome translocation t(1;11)(q23;p15) Blood. 1999;94(2):741–747. [PubMed] [Google Scholar]
- 163.Gervais C, Mauvieux L, Perrusson N, Helias C, Struski S, Leymarie V, Lioure B, Lessard M. A new translocation t(9;11)(q34;p15) fuses NUP98 to a novel homeobox partner gene, PRRX2, in a therapy-related acute myeloid leukemia. Leukemia. 2005;19(1):145–148. doi: 10.1038/sj.leu.2403565. [DOI] [PubMed] [Google Scholar]
- 164.Iwase S, Akiyama N, Sekikawa T, Saito S, Arakawa Y, Horiguchi-Yamada J, Yamada H. Both NUP98/TOP1 and TOP1/NUP98 transcripts are detected in a de novo AML with t(11;20)(p15;q11) Genes Chromosomes Cancer. 2003;38(1):102–105. doi: 10.1002/gcc.10239. [DOI] [PubMed] [Google Scholar]
- 165.Nebral K, Schmidt HH, Haas OA, Strehl S. NUP98 is fused to topoisomerase (DNA) IIbeta 180 kDa (TOP2B) in a patient with acute myeloid leukemia with a new t(3;11)(p24;p15) Clin Cancer Res. 2005;11(18):6489–6494. doi: 10.1158/1078-0432.CCR-05-0150. [DOI] [PubMed] [Google Scholar]
- 166.Jankovic D, Gorello P, Liu T, Ehret S, La Starza R, Desjobert C, Baty F, Brutsche M, Jayaraman PS, Santoro A, Mecucci C, Schwaller J. Leukemogenic mechanisms and targets of a NUP98/HHEX fusion in acute myeloid leukemia. Blood. 2008;111(12):5672–5682. doi: 10.1182/blood-2007-09-108175. [DOI] [PubMed] [Google Scholar]
- 167.Panagopoulos I, Kerndrup G, Carlsen N, Strombeck B, Isaksson M, Johansson B. Fusion of NUP98 and the SET binding protein 1 (SETBP1) gene in a paediatric acute T cell lymphoblastic leukaemia with t(11;18)(p15;q12) Br J Haematol. 2007;136(2):294–296. doi: 10.1111/j.1365-2141.2006.06410.x. [DOI] [PubMed] [Google Scholar]
- 168.Hussey DJ, Nicola M, Moore S, Peters GB, Dobrovic A. The (4;11)(q21;p15) translocation fuses the NUP98 and RAP1GDS1 genes and is recurrent in T-cell acute lymphocytic leukemia. Blood. 1999;94(6):2072–2079. [PubMed] [Google Scholar]
- 169.Ishikawa M, Yagasaki F, Okamura D, Maeda T, Sugahara Y, Jinnai I, Bessho M. A novel gene, ANKRD28 on 3p25, is fused with NUP98 on 11p15 in a cryptic 3-way translocation of t(3;5;11)(p25;q35;p15) in an adult patient with myelodysplastic syndrome/acute myelogenous leukemia. Int J Hematol. 2007;86(3):238–245. doi: 10.1532/IJH97.07054. [DOI] [PubMed] [Google Scholar]
- 170.von Lindern M, Fornerod M, van Baal S, Jaegle M, de Wit T, Buijs A, Grosveld G. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol Cell Biol. 1992;12(4):1687–1697. doi: 10.1128/mcb.12.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Soekarman D, von Lindern M, Daenen S, de Jong B, Fonatsch C, Heinze B, Bartram C, Hagemeijer A, Grosveld G. The translocation (6;9) (p23;q34) shows consistent rearrangement of two genes and defines a myeloproliferative disorder with specific clinical features. Blood. 1992;79(11):2990–2997. [PubMed] [Google Scholar]
- 172.von Lindern M, van Baal S, Wiegant J, Raap A, Hagemeijer A, Grosveld G. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3′ half to different genes: characterization of the set gene. Mol Cell Biol. 1992;12(8):3346–3355. doi: 10.1128/mcb.12.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Soman NR, Correa P, Ruiz BA, Wogan GN. The TPR-MET oncogenic rearrangement is present and expressed in human gastric carcinoma and precursor lesions. Proc Natl Acad Sci U S A. 1991;88(11):4892–4896. doi: 10.1073/pnas.88.11.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Greco A, Mariani C, Miranda C, Lupas A, Pagliardini S, Pomati M, Pierotti MA. The DNA rearrangement that generates the TRK-T3 oncogene involves a novel gene on chromosome 3 whose product has a potential coiled-coil domain. Mol Cell Biol. 1995;15(11):6118–6127. doi: 10.1128/mcb.15.11.6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Greil F, Moorman C, van Steensel B. DamID: mapping of in vivo protein-genome interactions using tethered DNA adenine methyltransferase. Methods Enzymol. 2006;410:342–359. doi: 10.1016/S0076-6879(06)10016-6. [DOI] [PubMed] [Google Scholar]
- 176.Schmid M, Durussel T, Laemmli UK. ChIC and ChEC; genomic mapping of chromatin proteins. Mol Cell. 2004;16(1):147–157. doi: 10.1016/j.molcel.2004.09.007. [DOI] [PubMed] [Google Scholar]