Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: biologic and clinical implications (original) (raw)
1 Introduction
One of the most important advances in tumor biology is the recognition that cancer is frequently driven by inherited or acquired alterations in specific gene(s) or their products [1, [2](/article/10.1007/s10555-015-9579-8#ref-CR2 "Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of Cancer: The Next Generation. Cell, 144(5), 646–674. doi: 10.1016/j.cell.2011.02.013
.")\]. Genomic alterations include changes in expression that can result from mutation, deletion, gene amplification, and/or translocation. Complicating matters, cancers often harbor multiple genetic alterations, but one or a few of these are thought to be primarily responsible for neoplastic behavior in any given tumor. These are the so-called “driver mutations,” while “passenger mutations” may have a more nuanced impact. A literature review suggests that over 1 % of human genes can be implicated as cancer drivers when they are mutated, with protein kinases comprising the largest subgroup of genes altered \[[3](/article/10.1007/s10555-015-9579-8#ref-CR3 "Greenman, C., Stephens, P., Smith, R., Dalgliesh, G. L., Hunter, C., Bignell, G., & Stratton, M. R. (2007). Patterns of somatic mutation in human cancer genomes. Nature, 446(7132), 153–158.")\].Among human signaling pathways, fibroblast growth factor (FGF)/fibroblast growth factor receptor (FGFR) is one of the pathways most enriched in non-synonymous mutations, including several candidate driver mutations [3]. A computational method designed to identify driver mutations within protein kinase datasets successfully identified multiple aberrations in the FGF/FGFR machinery [4]. In keeping with other genes implicated in neoplastic behavior, FGF/FGFR signaling is also involved in development, differentiation, growth, and survival mechanisms. Indeed, FGFR aberrations have been identified in both hereditary and neoplastic human diseases.
Most of the reported FGFR mutations that cause heritable human diseases are activating mutations which increase receptor signaling. These abnormalities are seen in craniofacial and skeletal syndromes such as the craniosynostoses [5–7] (Pfeiffer, Crouzon, Apert, Jackson-Weiss, Muenke, and Beare-Stevenson syndromes) and dwarfism syndromes [8, 9] (achondroplasia, thanatophoric dysplasia, and hypochondroplasia). The only reported inherited condition caused by loss of FGFR function is an autosomal dominant form of hereditary hypogonatotrophic hypogonadism 2 with or without anosmia [10, [11](/article/10.1007/s10555-015-9579-8#ref-CR11 " OMIM hypogonatotrophic hypogonadism 2. (n.d.). Retrieved from http://www.omim.org/entry/147950
.")\], which is caused by loss of function of FGFR1 \[[10](/article/10.1007/s10555-015-9579-8#ref-CR10 "Dodé, C., Teixeira, L., Levilliers, J., Fouveaut, C., Bouchard, P., Kottler, M.-L., & Hardelin, J.-P. (2006). Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS genetics, 2(10), e175."), [12](/article/10.1007/s10555-015-9579-8#ref-CR12 "Dodé, C., Levilliers, J., Dupont, J.-M., De Paepe, A., Le Dû, N., Soussi-Yanicostas, N., & Hardelin, J.-P. (2003). Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nature Genetics, 33(4), 463–465. doi:
10.1038/ng1122
."), [13](/article/10.1007/s10555-015-9579-8#ref-CR13 "Thurman, R. D., Kathir, K. M., Rajalingam, D., & Kumar, T. K. S. (2012). Molecular basis for the Kallmann syndrome-linked fibroblast growth factor receptor mutation. Biochemical and Biophysical Research Communications, 425(3), 673–678. doi:
10.1016/j.bbrc.2012.07.104
.")\] or a missense mutation in FGF8 \[[14](/article/10.1007/s10555-015-9579-8#ref-CR14 "Trarbach, E. B., Abreu, A. P., Silveira, L. F. G., Garmes, H. M., Baptista, M. T. M., Teles, M. G., & Latronico, A. C. (2010). Nonsense mutations in FGF8 gene causing different degrees of human gonadotropin-releasing deficiency. The Journal of Clinical Endocrinology and Metabolism, 95(7), 3491–3496. doi:
10.1210/jc.2010-0176
.")\]. Intriguingly, some of the same activating FGFR mutations seen in inherited syndromes are also seen in human cancers \[[14](/article/10.1007/s10555-015-9579-8#ref-CR14 "Trarbach, E. B., Abreu, A. P., Silveira, L. F. G., Garmes, H. M., Baptista, M. T. M., Teles, M. G., & Latronico, A. C. (2010). Nonsense mutations in FGF8 gene causing different degrees of human gonadotropin-releasing deficiency. The Journal of Clinical Endocrinology and Metabolism, 95(7), 3491–3496. doi:
10.1210/jc.2010-0176
."), [15](/article/10.1007/s10555-015-9579-8#ref-CR15 "Turner, N., & Grose, R. (2010). Fibroblast growth factor signalling: from development to cancer. Nature Reviews Cancer, 10(2), 116–129.")\]. Furthermore, FGF/FGFR aberrations reported in cancers are overwhelmingly thought to be gain-of-function changes, including gene amplifications and gene rearrangements \[[16](/article/10.1007/s10555-015-9579-8#ref-CR16 "Dienstmann, R., Rodon, J., Prat, A., Perez-Garcia, J., Adamo, B., Felip, E., & Tabernero, J. (2014). Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO, 25(3), 552–563. doi:
10.1093/annonc/mdt419
.")\].The goal of identification and characterization of driver mutations in cancer is, ultimately, to create successful anti-cancer therapies with which to prosecute these tumors; several such therapies already exist, demonstrating proof of principle [17, [18](/article/10.1007/s10555-015-9579-8#ref-CR18 "Yan, M., Parker, B. A., Schwab, R., & Kurzrock, R. (n.d.). HER2 aberrations in cancer: Implications for therapy. Cancer Treatment Reviews. doi: 10.1016/j.ctrv.2014.02.008
.")\]. Furthermore, for some gene targets, drugs may impact the course of cancer as well as non-malignant conditions that are driven by abnormalities in the cognate signal. JAK2 aberrations, for instance, are found in myelofibrosis, and JAK2 inhibitors such as ruxolitinib provide significant benefit in such patients \[[19](/article/10.1007/s10555-015-9579-8#ref-CR19 "Verstovsek, S., Mesa, R. A., Gotlib, J., Levy, R. S., Gupta, V., DiPersio, J. F., & Kantarjian, H. M. (2012). A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. New England Journal of Medicine, 366(9), 799–807. doi:
10.1056/NEJMoa1110557
.")\]. At the same time, the JAK2 inhibitor tofacitinib can benefit patients with rheumatoid arthritis and is approved for that indication \[[20](/article/10.1007/s10555-015-9579-8#ref-CR20 "Lee, E. B., Fleischmann, R., Hall, S., Wilkinson, B., Bradley, J. D., Gruben, D., & van Vollenhoven, R. F. (2014). Tofacitinib versus Methotrexate in Rheumatoid Arthritis. New England Journal of Medicine, 370(25), 2377–2386. doi:
10.1056/NEJMoa1310476
.")\]. In the case of FGF/FGFR, multiple drugs targeting this pathway have entered the clinic \[[16](/article/10.1007/s10555-015-9579-8#ref-CR16 "Dienstmann, R., Rodon, J., Prat, A., Perez-Garcia, J., Adamo, B., Felip, E., & Tabernero, J. (2014). Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO, 25(3), 552–563. doi:
10.1093/annonc/mdt419
.")\]. Herein, we discuss the landscape of diseases that are driven by aberrant FGF/FGFR machinery.2 Molecular biology of FGF/FGFR signaling
Fibroblast growth factors (FGFs) and their receptors (FGFRs) are evolutionarily conserved transmembrane growth factor receptors with wide tissue distribution in all vertebrates. FGFs and FGFRs share homologies among their respective groups and with other signaling molecules. FGFRs in particular are similar to other signaling receptors, including vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), and other receptor tyrosine kinases [21]. However, there are important differences between the individual signaling molecules that allow for precise control of a full range of processes, including development, cell survival, differentiation, motility, angiogenesis, and carcinogenesis.
2.1 Receptors
In humans, there are five known FGFRs, called FGFR1–FGFR4 and FGFRL1 (also known as FGFR5). FGFR1–FGFR4 are typical growth factor receptor tyrosine kinases, with extracellular immunoglobulin (Ig)-like domains and intracellular tyrosine kinase domains, while FGFRL1 lacks the intracellular kinase domain and has less clear function(s)[22, 23]. Upon binding of their ligands, the typical receptors homo- or hetero-dimerize, leading to sequential phosphorylation of specific intracellular tyrosine residues and activation of an intracellular signaling cascade and gene transcription [24] (Fig. 1). The FGFR signaling pathway interacts with several other important intracellular pathways, including PI3K/Akt, Wnt, hedgehog, and bone morphogenic protein (BMP) [24, 25]. FGFR1–4 have different ligand specificities based on developmental aspects, tissue distribution, and RNA splicing variation [26]. For example, the FGFR2b isoform is predominantly expressed in epithelial cells, while the FGFR2c isoform is expressed predominantly in mesenchymal cells, and switching from FGFR2b to FGFR2c occurs during progression and invasion of prostate and bladder cancers [27].
Fig. 1
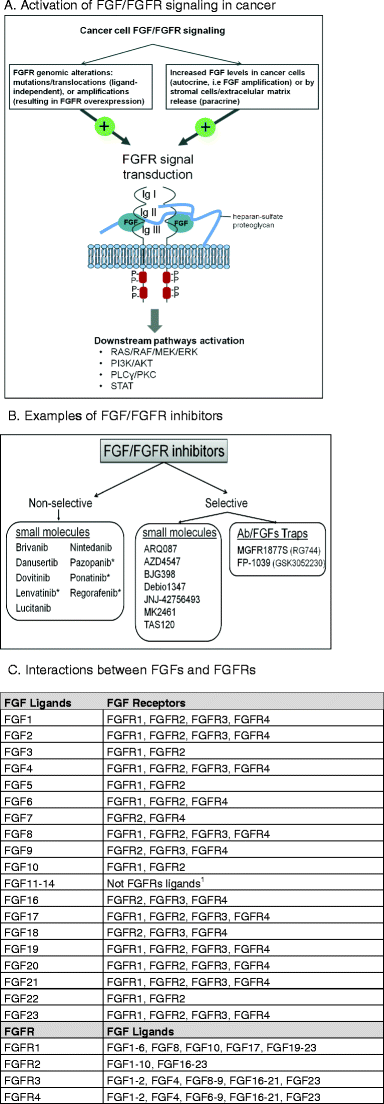
FGF/FGFR activation in cancer and inhibitors. a The activation of FGF/FGFR signaling in cancer. The structure of FGF/FGFR comprises two receptor molecules, two FGFs, and one heparan sulfate proteoglycan chain. The FGFRs are formed by three immunoglobulin domains (IgI–III), a transmembrane helix, and intracellular tyrosine kinase domains. The mechanisms driving FGF signaling in cancer can be divided into two categories: first, genomic alterations of FGFR that can lead to ligand-independent receptor signaling and, second, alterations that support a ligand-dependent signaling activation. Following FGF binding to FGFR and heterodimerization, the tyrosine kinase domains phosphorylate each other, leading to the activation of key downstream pathways. b Examples of FGF/FGFR inhibitors; asterisk denotes FDA-approved drugs in cancer; Ab = Antibody; FP-1039 (GSK3052230) is a ligand trap, i.e., sequesters FGFs and inhibits their signaling [[71](/article/10.1007/s10555-015-9579-8#ref-CR71 "Bellovin, D. I., Palencia, S., Hestir, K., Lee, E., DeYoung, M. P., Brennan, T., & Baker, K. (2014). Abstract 5449: FP-1039/GSK3052230, an FGF ligand trap, enhances VEGF antagonist therapy in preclinical models of RCC and HCC. Cancer Research, 74(19 Supplement), 5449–5449. doi: 10.1158/1538-7445.AM2014-5449
.")\]. **c** The interactions between FGFs and FGFRs; references: Guillemot et al. \[[32](/article/10.1007/s10555-015-9579-8#ref-CR32 "Guillemot, F., & Zimmer, C. (2011). From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron, 71(4), 574–588. doi:
10.1016/j.neuron.2011.08.002
.")\], Powers et al. \[[191](/article/10.1007/s10555-015-9579-8#ref-CR191 "Powers, C. J., McLeskey, S. W., & Wellstein, A. (2000). Fibroblast growth factors, their receptors and signaling. Endocrine-Related Cancer, 7(3), 165–197.")\], Ornitz et al. \[[192](/article/10.1007/s10555-015-9579-8#ref-CR192 "Itoh, N., & Ornitz, D. M. (2004). Evolution of the Fgf and Fgfr gene families. Trends in Genetics, 20(11), 563–569. doi:
10.1016/j.tig.2004.08.007
.")\], Zhang et al. \[[34](/article/10.1007/s10555-015-9579-8#ref-CR34 "Zhang, X., Ibrahimi, O. A., Olsen, S. K., Umemori, H., Mohammadi, M., & Ornitz, D. M. (2006). Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. The Journal of biological chemistry, 281(23), 15694–15700.")\]. Interaction between FGF ligands and receptors is an evolving field; variability may be observed between studies and tissue types. 1FGF11-14 are not ligands for FGFRs and are known as FGF homologous factors (FHF1–4)\[[28](/article/10.1007/s10555-015-9579-8#ref-CR28 "Olsen, S. K., Garbi, M., Zampieri, N., Eliseenkova, A. V., Ornitz, D. M., Goldfarb, M., & Mohammadi, M. (2003). Fibroblast Growth Factor (FGF) Homologous Factors Share Structural but Not Functional Homology with FGFs. Journal of Biological \ldots.
"), [29](/article/10.1007/s10555-015-9579-8#ref-CR29 "Itoh, N., & Ornitz, D. M. (2008). Functional evolutionary history of the mouse Fgf gene family. Developmental dynamics : an official publication of the American Association of Anatomists, 237(1), 18–27.")\]. There is no human FGF152.2 Ligands
There are 18 human ligands for FGFRs (Fig. 1). They are FGF1 (acidic FGF), FGF2 (basic FGF), FGF3, FGF4, FGF5, FGF6, FGF7 (KGF), FGF8, FGF9, FGF10, FGF16, FGF17, FGF18, FGF19, FGF20, FGF21, FGF22, and FGF23. FGF11–14 are not ligands for FGFRs and are known as FGF homologous factors (FHF1–4) [28, 29]. There is no human FGF15 (FGF15 is the mouse equivalent of human FGF19) [30]. The FGFR ligands are secreted proteins that bind to the extracellular matrix, thereby restricting their influence to the tissue where they are produced (autocrine or paracrine function). However, three of the FGFs—FGF19, FGF21, and FGF23—bind less tightly to extracellular matrix heparin sulfates, so they are able to act systemically in an endocrine fashion (hormone-like), allowing them to spread from their production site into the circulation [30, 31]. Most FGFs are secreted proteins with cleavable amino terminal portions, but FGF9, FGF16, and FGF20 have non-cleavable secretion sequences, and FGF1 and FGF2 have no secretion sequences, although they are found in the extracellular compartment, suggesting an alternate process of release [29, [32](/article/10.1007/s10555-015-9579-8#ref-CR32 "Guillemot, F., & Zimmer, C. (2011). From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron, 71(4), 574–588. doi: 10.1016/j.neuron.2011.08.002
.")\]. Most FGFs are released from binding to the extracellular matrix via action of heparin sulfate proteoglycans and/or fibroblast growth factor–binding protein (FGFBP1)\[[33](/article/10.1007/s10555-015-9579-8#ref-CR33 "Abuharbeid, S., Czubayko, F., & Aigner, A. (2006). The fibroblast growth factor-binding protein FGF-BP. The international journal of biochemistry & cell biology, 38(9), 1463–1468.")\].2.3 FGF/FGFR interaction and function
In general, FGFs are promiscuous, and most can bind to any of the four main FGFRs, although some of the FGF/FGFR ligand/receptor pairs differ in their affinities (Fig. 1c). FGFs interact with cell surface heparan sulfate proteoglycans (HSPGs) and with the transmembrane protein Klotho (for the hormone-like FGF19, 20, 23) to stabilize binding to FGFRs. Ornitz et al. [34, 35] systemically investigated affinities of FGFs for each FGFR isoform and showed, for example, that FGFR2b is a high-affinity receptor for FGF1, FGF7, FGF10, and FGF22, while FGFR2c has high affinity for FGF1, FGF2, FGF4, FGF6, FGF8, FGF9, and FGF20. But, differential expression of either FGFs or FGFRs in time or tissue may also contribute to tissue-specific effects of FGF/FGFR signaling. For example, FGF1 and FGF2 are expressed in both embryonic and adult tissues, while FGF7–FGF9 are predominantly expressed in developing or in restricted adult tissues [36].
The differences in activity/function of the different FGF/FGFR pairs are also highlighted by murine gene knockout experiments. Mice heterozygous for FGFR knockout mutations develop normally, so haplo-insufficiency is not likely to be a factor [37]. However, mice homozygous for FGFR1 or FGFR2 null mutations die in utero, and FGFR3-null mice develop normally other than overgrowth of cancellous bones and deafness [37]. Most of the FGFs have also been knocked out in mouse models, with varying phenotypic effects, including lethality (FGF4, FGF8, FGF9, FGF10, FGF19, FGF18, and FGF23), defects in neuromusculoskeletal development or function (FGF2, FGF3, FGF6, FGF7, FGF12, FGF14, FGF17, FGF18), cardiac defects (FGF2, FGF9, FGF16, FGF19), and no identifiable abnormality (FGF1) [29].
2.4 Activation of signaling
To signal, FGFs have to be released from the extracellular matrix by heparinases, proteases, or specific fibroblast growth factor–binding protein (FGFBP1). FGFBP1 are secreted heparin proteins that reversibly bind FGF1 and FGF2, releasing them from the extracellular matrix and increasing the local levels of free ligand available for receptor binding [38]. FGFBP1 is secreted by keratinocytes and human epidermal carcinomas, particularly squamous cell carcinomas [33, 38]. Its expression is seen in development, wound healing, cancer, and angiogenesis, and it is downregulated by pharmacologic agents in vitro [33]. The fibronectin-leucine-rich transmembrane protein 3 (FLRT3) is co-expressed with FGF8 during development, co-immunoprecipitates with FGF8/FGFRs, physically interacts with FGFRs via its fibronectin type III domain, and increases activity through the MAPK pathway, facilitating FGFR signaling [39, 40].
2.5 Inhibition of signaling
FGF/FGFR signaling is negatively regulated by several mechanisms. FGFR stimulation activates sprouty proteins (SPRY1–4), which in turn negatively feedbacks on FGF/FGFR/MAPK signaling by interacting with growth factor receptor bound protein 2 (GRB2), son of sevenless homolog 1 (SoS1), and/or RAF proto-oncogene serine/threonine-protein kinase (RAF1) [41, 42]. Intriguingly, sprouty proteins are thought to be general inhibitors of receptor tyrosine kinase growth factor signaling via inhibition of Ras-MAPK signaling, but their behavior differs among specific growth factor receptors. For example, sprouty protein inhibits FGFR signaling but potentiates epidermal growth factor receptor (EGFR) signaling [43]. The cytoplasmic domain of similar expression to FGF (SEF, also known as IL-17 receptor D) interacts with the cytoplasmic domain of FGFR and inhibits downstream signaling of the pathway [43]. Finally, MAP kinase phosphatases (MKPs) dephosphorylate ERKs and thereby downregulate the signaling pathway. MKP3 functions within the cytoplasm, whereas MKP1 is localized in the nucleus [43].
FGFRL1 is the atypical receptor that lacks an intracellular kinase domain. Like the other FGFRs, it is found in all vertebrates, is expressed in a wide variety of tissues, and binds to some FGF ligands. In vitro experiments show that FGFRL1 binds FGF3, FGF4, FGF8, FGF10, and FGF22, but not the other FGFs tested [44]. Unlike FGFR1–4, it is shed from cell membranes [44], suggesting that it may serve as a ligand trap to negatively regulate signaling [45]. Other regulatory mechanisms might include heterodimerization with other FGFRs to prevent trans-autophophorylation and thereby negatively regulating intracellular signaling or by increasing membrane turnover rates of the other FGFRs, but there is currently no evidence that these mechanisms exist [45]. The exact functions of FGFRL1 are not known. FGFRL1-null mice die in infancy with diaphragmatic defects and renal agenesis [45], and there are case reports of FGFRL1 mutations in human disease: craniosynostosis [46] and ovarian cancer [47].
3 FGF/FGFR aberrant signaling in human disease
3.1 Inherited syndromes
Several types of inherited syndromes are due to germline aberrations in FGFR. These include craniosynostosis syndromes and achondroplasia [[48](/article/10.1007/s10555-015-9579-8#ref-CR48 "Garcia, S., Dirat, B., Tognacci, T., Rochet, N., Mouska, X., Bonnafous, S., & Gouze, E. (2013). Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Science Translational Medicine, 5(203), 203ra–124. doi: 10.1126/scitranslmed.3006247
.")\], mainly due to gain-of-function mutations, as well as loss-of-function anomalies associated with congenital hypogonadotropic hypogonadism (Table [1](/article/10.1007/s10555-015-9579-8#Tab1)). Interestingly, although some of these aberrations are identical to those that, in somatic form, are associated with cancer, for the most part, the individuals with these hereditary syndromes have not been reported to have an increased incidence of cancer. Apart from case reports \[[49](/article/10.1007/s10555-015-9579-8#ref-CR49 "Cohen, M. M. (2003). Neoplasms associated with alterations in fibroblast growth factor receptors. American journal of medical genetics Part A, 119A(2), 97–100.")–[53](/article/10.1007/s10555-015-9579-8#ref-CR53 "Bourdeaut, F., Miquel, C., Di Rocco, F., Grison, C., Richer, W., Brugieres, L., & Collet, C. (2013). Germline mutations in FGF receptors and medulloblastomas. American journal of medical genetics Part A, 161A(2), 382–385.")\], there is no epidemiologic evidence that people with craniosynostosis, dwarfism, or congenital hypogonadotropic hypogonadism are at increased risk for malignancy. This is possibly related to differential effects of activating FGFR mutations in cell or tissue type in the context of stage of development. It is possible, for instance, that there are as yet unknown anti-neoplastic compensatory effects in individuals with these disorders, perhaps stemming from the presence of germline FGFR aberrations during development as opposed to the appearance of somatic FGFR mutations in human cancers.Table 1 Examples of FGFR aberrations in inherited syndromes
3.2 Somatic aberrations in benign conditions
Somatic or acquired mutations in FGFR3 have been observed in benign skin conditions like seborrheic keratosis and epidermal nevi [54, 55]. These aberrations are activating and, when evaluated, are not seen in adjacent normal skin [55]. Many of these are identical to mutations that are also seen in bladder and/or cervical cancers.
3.3 Somatic aberrations in malignancies
Aberrations in FGFR and its ligands are common in malignancy (Tables 2, 3, and 4 and Figs. 2 and 3). Across malignancies, FGF anomalies are found in ≈14 % and FGFR in ≈7 % of malignancies [[56](/article/10.1007/s10555-015-9579-8#ref-CR56 "cbioportal. (n.d.). Retrieved from http://www.cbioportal.org/public-portal/index.do
.")\] (FGF/FGFR in about 20 %).Table 2 Specific examples of FGFR alterations in cancer
Table 3 Examples of FGFR mutations and copy number alterations in cancer
Table 4 Examples of FGF ligand mutations and copy number aberrations in cancer
Fig. 2
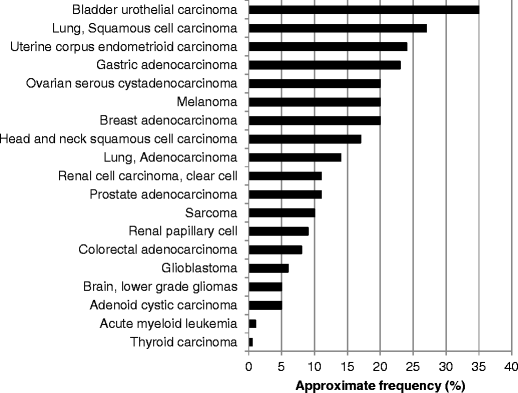
Approximate frequencies of FGFR alterations in diverse cancers. Data was extracted/analyzed based on cbioportal at http://www.cbioportal.org/public-portal (accessed November 2014). Most of the studies included >200 patients. Alterations in FGFR1, FGFR2, FGFR3, and FGFR4 were included. Please refer to Table 3 for more details and additional references. Abbreviations: FGFR = fibroblast growth factor receptor
Fig. 3
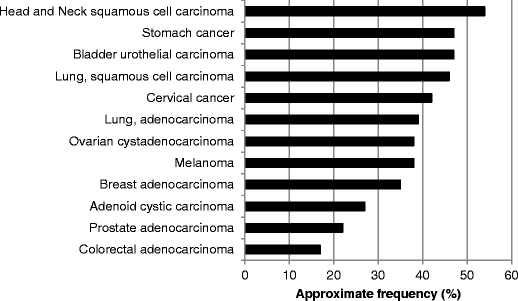
Approximate frequencies of FGF ligand alterations in diverse cancers. Data was extracted/analyzed based on data from cbioportal at http://www.cbioportal.org/public-portal (accessed November 2014). Cervical cancer included squamous cell carcinoma and endocervical adenocarcinoma. Please refer to Table 4 for more details and additional references. Abbreviations: FGF = fibroblast growth factor
3.3.1 FGFR alterations
While almost any FGFR genes can be altered in many cancer types, some acquired genetic aberrations are more striking in prevalence or in cancer type(s) than others. The most common abnormalities reported to date are gene amplifications of FGFR1–3 (Table 2). These are generally assumed to represent activation or gain-of-function amplifications, but full molecular characterization of the effects of gene amplification within specific cellular or cancer microenvironment contexts is not fully available. The cancers in which FGFR gene amplifications are most frequent include bladder urothelial carcinomas (FGFR1), squamous cell lung cancer (FGFR1), head and neck squamous cell cancer (FGFR1), uterine carcinosarcoma (FGFR3), breast adenocarcinoma (FGFR1), and gastric adenocarcinoma (FGFR2) (Tables 2 and 3, and Fig. 2). In fact, FGFR2 was first identified in a gastric cancer cell line, and it is preferentially amplified/overexpressed in the diffuse type of gastric cancer, which correlates with poor prognosis, at least in a Japanese population [57]. More commonly, however, mutations in FGFR3 characterize bladder carcinoma. Indeed, activating mutations of fibroblast growth factor receptor-3 (FGFR3) have been described in approximately 75 % of low-grade papillary bladder tumors. In muscle-invasive disease, FGFR3 mutations are found in 20 % of tumors, but overexpression of FGFR3 is observed in about half of cases [58].
Loss-of-function alterations are relatively uncommon in cancer (a pattern that is also seen in hereditary disorders). However, there is at least one report indicating that some melanoma cell lines demonstrate FGFR2 loss of function [59].
FGFR1 amplification is prevalent in both squamous cell carcinoma of the lung and of the head and neck, suggesting a possible common underlying mechanism of carcinogenesis in these smoking-related carcinomas. Perhaps more importantly, development of FGFR1 inhibitors represents a viable targeted therapy for use in squamous cell lung cancers [60, 61].
3.3.2 FGF alterations
FGF abnormalities are for the most part amplifications (Fig. 3 and Table 4). There are few reported data regarding the mechanism(s) of action of these amplifications. Nevertheless, it seems reasonable to hypothesize that abundance of ligand could lead to increased receptor signaling, but the reality may be more complex, perhaps relating to the stoichiometric proportions of the different ligands present. Of note, three of the ligands, FGF3, FGF4, and FGF19, are frequently co-amplified on 11q13 [62]. This amplicon is present in several cancers, including breast, bladder, and squamous cell carcinoma of the lung and head and neck (Fig. 3 and Table 4).
3.3.3 FGFR rearrangements
Chromosomal translocation gives rise to chimeric gene products with aberrant function (Table 2). In general, fusion gene proteins result from the fusion of a “partner gene” with a tyrosine kinase domain derived from the FGFR family member gene. As a result of their constant dimerization state, they are constitutively active in the absence of ligand. The FGFR1 gene can be fused to other genes including BCR, ZNF198, CEP110, FGFR1OP1, FGFR1OP2, HERVK, TRIM24, LRRFIP1, and MYO18A, in the 8p11 myeloproliferative syndrome manifested by myeloproliferative neoplasms and peripheral blood eosinophilia without basophilia [[63](/article/10.1007/s10555-015-9579-8#ref-CR63 "Jackson, C. C., Medeiros, L. J., & Miranda, R. N. (2010). 8p11 myeloproliferative syndrome: a review. Human Pathology, 41(4), 461–476. doi: 10.1016/j.humpath.2009.11.003
.")\]; it is fused to the _FOXO1_ gene in alveolar rhabdomyosarcoma, and the _FOXO1-FGFR1_ fusion gene is amplified \[[64](/article/10.1007/s10555-015-9579-8#ref-CR64 "Liu, J., Guzman, M. A., Pezanowski, D., Patel, D., Hauptman, J., Keisling, M., & de Chadarévian, J.-P. (2011). FOXO1-FGFR1 fusion and amplification in a solid variant of alveolar rhabdomyosarcoma. Modern Pathology: An Official Journal of the United States and Canadian Academy of Pathology, Inc, 24(10), 1327–1335. doi:
10.1038/modpathol.2011.98
.")\]. The _FGFR3_ gene is fused to the _MMSET_ gene as a result of a t(4;14)(p16.3;q32) chromosomal translocation in 10–20 % of multiple myeloma \[[65](/article/10.1007/s10555-015-9579-8#ref-CR65 "Grand, E. K., Chase, A. J., Heath, C., Rahemtulla, A., & Cross, N. C. P. (2004). Targeting FGFR3 in multiple myeloma: inhibition of t(4;14)-positive cells by SU5402 and PD173074. Leukemia, 18(5), 962–966. doi:
10.1038/sj.leu.2403347
.")\]; it is fused to the _ETV6_ gene in peripheral T cell lymphoma with a t(4;12)(p16;p13) chromosomal translocation \[[66](/article/10.1007/s10555-015-9579-8#ref-CR66 "Yagasaki, F., Wakao, D., Yokoyama, Y., Uchida, Y., Murohashi, I., Kayano, H., & Bessho, M. (2001). Fusion of ETV6 to fibroblast growth factor receptor 3 in peripheral T-cell lymphoma with a t(4;12)(p16;p13) chromosomal translocation. Cancer Research, 61(23), 8371–8374.")\]. In glioblastoma multiforme, _FGFR1_ and _FGFR3_ genes are fused to neighboring _TACC1_ and _TACC3_ genes due to interstitial deletions, respectively \[[67](/article/10.1007/s10555-015-9579-8#ref-CR67 "Katoh, M., & Katoh, M. (2003). Recombination cluster around FGFR2-WDR11-HTPAPL locus on human chromosome 10q26. International Journal of Molecular Medicine, 11(5), 579–583."), [68](/article/10.1007/s10555-015-9579-8#ref-CR68 "Singh, D., Chan, J. M., Zoppoli, P., Niola, F., Sullivan, R., Castano, A., & Iavarone, A. (2012). Transforming Fusions of FGFR and TACC Genes in Human Glioblastoma. Science, 337(6099), 1231–1235.")\].4 FGF/FGFR signaling inhibitors and cancer therapy
4.1 FDA approved drugs that target FGFR
Only four drugs (ponatinib, regorafenib, pazopanib, and more recently lenvatinib) are FDA-approved for use in human cancers (Table 5). Ponatinib is a multi-tyrosine kinase inhibitor that was approved for imatinib-resistant chronic myelogenous leukemia (CML) and Philadelphia chromosome-positive (CP) acute lymphoblastic leukemia (ALL). The efficacy results demonstrated a 54 % major cytogenetic response (MCyR) rate in patients with CP-CML, and seventy percent of patients with CP-CML with the T315I mutation in BCR-ABL achieved MCyR. In addition of targeting BCR-ABL, ponatinib can also inhibit members of the VEGFR, PDGFR, FGFR (IC50 = 2 nM for FGFR1), and SRC families of kinases, KIT, or RET, with IC50 between 0.1 and 20 nM. Of note, ponatinib was briefly taken off the market by the FDA because of concerns about cardiovascular side effects (clotting), but soon after, it was returned to the market with updated safety monitoring recommendations. Regorafenib was approved for imatinib-resistant gastrointestinal stromal tumor (GIST) and metastatic colorectal cancer, based on a statistically significant survival prolongation observed in patients randomized to receive regorafenib (6.4 vs 5.0 months in the placebo arm, P = 0.01). Regorafenib, and its active metabolites inhibit multiple membrane-bound and intracellular kinases including those in the RET, VEGFR1, VEGFR2, VEGFR3, KIT, PDGFR-α and -β, FGFR1-2, and Abl pathways. Pazopanib was approved for advanced renal cell carcinoma (based on a progression-free survival of 9.2 months compared to 4.2 months in the placebo arm), as well as soft tissue sarcomas (improved progression-free survival: 4.6 months versus 1.6 for patients who received placebo). Pazopanib is a multi-tyrosine kinase inhibitor whose targets include VEGFR1-3, PDGFR-α and -β, FGFR-1 and -3, and KIT. Lastly, lenvatinib is a multi-kinase inhibitor (targets including VEGFR1–3, FGFR1–4, PDGFR-α, KIT, and RET) indicated for the treatment of patients with locally recurrent or metastatic, progressive, radioactive iodine-refractory differentiated thyroid cancer. The recent approval in February 2015 was based on an improved progression-free survival (18.3 vs 3.6 months in the placebo group, P < 0.001). The approvals of these four agents (ponatinib, regorafenib, pazopanib, and lenvatinib), all three of which are multi-kinase inhibitors, were not based on activity against FGFR. Of interest, nintedanib is an FGFR inhibitor that is FDA-approved for a non-cancer indication—idiopathic pulmonary fibrosis (Table 5).
Table 5 Examples of drugs that inhibit FGF/FGFR pathway signaling
4.2 Experimental agents that target FGF/FGFR
There are several pharmacologic agents that have been or are being developed for inhibition of FGFR via targeting of the ATP binding site of the intracellular tyrosine kinase domain(s) (Table 5). The inhibition varies by their affinities for FGFR signaling, as most of these molecules are promiscuous kinase inhibitors (inhibiting VEGF, PDGF, and many other TKIs in addition to FGFR). Figure 1b gives examples of selective versus non-selective FGFR inhibitors. Of note, the dual kinase inhibitor (VEGFR/FGFR) lucitanib has shown activity in FGFR1-amplified breast cancer, with an overall response rate of 50 % [[69](/article/10.1007/s10555-015-9579-8#ref-CR69 "Soria, J.-C., DeBraud, F., Bahleda, R., Adamo, B., Andre, F., Dientsmann, R., & Tabernero, J. (2014). Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO, 25(11), 2244–2251. doi: 10.1093/annonc/mdu390
.")\]. There are also selective tyrosine kinase inhibitors (TKIs) available (Table [5](/article/10.1007/s10555-015-9579-8#Tab5) and Fig. [1b](/article/10.1007/s10555-015-9579-8#Fig1)). While several of these agents are currently in clinical trials, none of the more highly selective FGFR inhibitors (e.g., BGJ398 or AZD4547) are currently FDA-approved. A recent study demonstrated that Debio 1347 (a selective orally available FGFR1–3 inhibitor) displayed preferential anti-tumor activity against cells with FGFR genetic alterations in a panel of 327 cancer cell lines and xenograft models \[[70](/article/10.1007/s10555-015-9579-8#ref-CR70 "Nakanishi, Y., Akiyama, N., Tsukaguchi, T., Fujii, T., Sakata, K., Sase, H., & Ishii, N. (2014). The Fibroblast Growth Factor Receptor Genetic Status as a Potential Predictor of the Sensitivity to CH5183284/Debio 1347, a Novel Selective FGFR Inhibitor. Molecular Cancer Therapeutics, 13(11), 2547–2558. doi:
10.1158/1535-7163.MCT-14-0248
.")\]. Debio 1347 is currently under investigation for the treatment of patients harboring FGFR genetic alterations.Of interest, other types of agents have been developed. As an example, FP-1039 is a soluble fusion protein, consisting of the extracellular domains of human FGFR1 linked to the Fc region of the human immunoglobulin G1; it is designed to bind multiple FGF ligands [[71](/article/10.1007/s10555-015-9579-8#ref-CR71 "Bellovin, D. I., Palencia, S., Hestir, K., Lee, E., DeYoung, M. P., Brennan, T., & Baker, K. (2014). Abstract 5449: FP-1039/GSK3052230, an FGF ligand trap, enhances VEGF antagonist therapy in preclinical models of RCC and HCC. Cancer Research, 74(19 Supplement), 5449–5449. doi: 10.1158/1538-7445.AM2014-5449
.")\] (TRAP molecule).A large number of other drugs and indications are being pursued. Some FGFR inhibitors have failed to meet their phase III endpoints (Table 5). The majority of the failed trials, however, have been performed in patient populations that were not biomarker-selected.
4.3 Resistance mechanisms
In a recent phase 1 study reporting on patients with FGFR1-amplified (identified by fluorescent/chromogenic in situ hybridization) advanced or metastatic lung squamous cell carcinoma (SCC) treated with the selective pan-FGFR inhibitor BGJ398, only a limited number of patients achieved relatively short-lived partial responses (2 of 17 patients, 11.7 %); responses lasted 3 and 8 months. These observations suggest the existence of resistance mechanisms. Sohl et al. [[72](/article/10.1007/s10555-015-9579-8#ref-CR72 "Sohl, C. D., Ryan, M. R., Luo, B., Frey, K. M., & Anderson, K. S. (2015). Illuminating the Molecular Mechanisms of Tyrosine Kinase Inhibitor Resistance for the FGFR1 Gatekeeper Mutation: The Achilles’ Heel of Targeted Therapy. ACS chemical biology. doi: 10.1021/acschembio.5b00014
.")\] reported that resistance mutations at the “gatekeeper” residue may arise (FGFR1 V561M mutation confers a 38-fold increase in autophosphorylation and significant resistance to lucitanib), leading to tumor progression and explaining the non-durable responses. For instance, it has also been shown that the heterozygous gatekeeper mutation FGFR3 V555M appeared as a mechanism of acquired resistance to selective FGFR inhibitors \[[73](/article/10.1007/s10555-015-9579-8#ref-CR73 "Chell, V., Balmanno, K., Little, A. S., Wilson, M., Andrews, S., Blockley, L., & Cook, S. J. (2013). Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene, 32(25), 3059–3070. doi:
10.1038/onc.2012.319
.")\]. Several other activating mutations were identified in FGFR2-expressing cells treated with high concentrations of dovitinib, and the multi-kinase inhibitor ponatinib inhibitory activity was affected by the V565I gatekeeper mutation \[[74](/article/10.1007/s10555-015-9579-8#ref-CR74 "Byron, S. A., Chen, H., Wortmann, A., Loch, D., Gartside, M. G., Dehkhoda, F., & Pollock, P. M. (2013). The N550K/H mutations in FGFR2 confer differential resistance to PD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neoplasia (New York, N.Y.), 15(8), 975–988.")\]. In addition, a previously undescribed FGFR3 variant was identified as a key contributor to resistance in the MGH156-1A cell line derived from a patient with acquired resistance to EGFR TKIs \[[75](/article/10.1007/s10555-015-9579-8#ref-CR75 "Crystal, A. S., Shaw, A. T., Sequist, L. V., Friboulet, L., Niederst, M. J., Lockerman, E. L., & Engelman, J. A. (2014). Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science, 346(6216), 1480–1486. doi:
10.1126/science.1254721
.")\], and follow-up studies clearly indicated that FGFR inhibitors re-sensitized these cells to EGFR inhibitors.Besides secondary mutations in the kinase domain, resistance to FGFR kinase inhibitors may also occur through activation of alternative signaling pathways, as demonstrated by Harbinski et al. [[76](/article/10.1007/s10555-015-9579-8#ref-CR76 "Harbinski, F., Craig, V. J., Sanghavi, S., Jeffery, D., Liu, L., Sheppard, K. A., & Tiedt, R. (2012). Rescue screens with secreted proteins reveal compensatory potential of receptor tyrosine kinases in driving cancer growth. Cancer Discovery, 2(10), 948–959. doi: 10.1158/2159-8290.CD-12-0237
.")\] who showed a broad and versatile potential for tyrosine kinase receptor from the FGFR, HER, and MET family to compensate for each other. Javidi-Sharifi et al. \[[77](/article/10.1007/s10555-015-9579-8#ref-CR77 "Javidi-Sharifi, N., Traer, E., Martinez, J., Gupta, A., Taguchi, T., Dunlap, J., & Tyner, J. W. (2015). Crosstalk between KIT and FGFR3 Promotes Gastrointestinal Stromal Tumor Cell Growth and Drug Resistance. Cancer Research, 75(5), 880–891. doi:
10.1158/0008-5472.CAN-14-0573
.")\] suggest that some patients with gastrointestinal stromal tumor (GIST) treated with imatinib can develop a functional dependence on FGFR3, illustrated by the fact that the addition of the FGFR3 ligand FGF2 to GIST cells restored KIT phosphorylation during imatinib treatment. Furthermore, signaling crosstalk between KIT and FGFR3 activated the MAPK pathway to promote resistance to imatinib. _FGFR_ amplification and overexpression have also been related to poor prognosis and endocrine resistance in breast cancer \[[78](/article/10.1007/s10555-015-9579-8#ref-CR78 "Turner, N., Pearson, A., Sharpe, R., Lambros, M., Geyer, F., Lopez-Garcia, M. A., & Ashworth, A. (2010). FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer research, 70(5), 2085–2094. doi:
10.1158/0008-5472.CAN-09-3746
.")\]. Of note, relationships between cyclins and FGF/FGFRs have also been reported at the protein level. For instance, a study showed that FGFR4 contributed to the maintenance of CCND1 via the mTOR translation pathway, and several other studies demonstrated cooperation between FGFR and CCND1 \[[79](/article/10.1007/s10555-015-9579-8#ref-CR79 "Schwaederlé, M., Daniels, G. A., Piccioni, D. E., Fanta, P. T., Schwab, R. B., Shimabukuro, K. A., … Kurzrock, R. (2014). Cyclin alterations in diverse cancers: Outcome and co-amplification network. Oncotarget.
")\]. Finally, most patients with advanced cancer have complex molecular portfolios, and hence there may be multiple genomic drivers that are active and supplant the role of FGFR \[[80](/article/10.1007/s10555-015-9579-8#ref-CR80 "Wheler, J. J., Parker, B. A., Lee, J. J., Atkins, J. T., Janku, F., Tsimberidou, A. M., … Kurzrock, R. (2014). Unique molecular signatures as a hallmark of patients with metastatic breast cancer: Implications for current treatment paradigms. Oncotarget.
")–[82](/article/10.1007/s10555-015-9579-8#ref-CR82 "Tsimberidou, A.-M., Iskander, N. G., Hong, D. S., Wheler, J. J., Falchook, G. S., Fu, S., & Kurzrock, R. (2012). Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clinical cancer research: an official journal of the American Association for Cancer Research, 18(22), 6373–6383. doi:
10.1158/1078-0432.CCR-12-1627
.")\]. In that context, it appears evident that identification of resistance mechanisms is crucial to crafting effective drug combinations.5 Conclusion
The FGF/FGFR pathway is crucial to a variety of human diseases. There are five known FGFRs, FGFR1–FGFR4 and FGFRL1, and 18 human ligands for FGFRs. FGFR germline mutations (activating) can cause skeletal disorders, primarily dwarfism (generally mutations in FGFR3) and craniofacial malformation syndromes (usually mutations in FGFR1 and FGFR2). Loss-of-function mutations in FGF signaling are seen in congenital hypogonadotropic hypogonadism (including the Kallman syndrome variant with anosmia). Interestingly, many of the aberrations that cause the inherited skeletal disorders are also seen in human cancers.
The most common abnormalities in malignancies are gene amplifications of FGFR1–3 or of the FGF ligands. The cancers in which FGFR gene amplifications are most frequent include squamous cell lung cancer (FGFR1), head and neck squamous cell cancer (FGFR1), bladder (transitional cell) cancer (FGFR1), endometrial cancer (FGFR1), gastric adenocarcinoma (FGFR2), breast adenocarcinoma (FGFR1), and prostate adenocarcinoma (FGFR1). Point mutations are seen in all FGFRs but are less frequent in FGFs. For instance, mutations in FGFR3 are frequent in bladder carcinoma, and FGFR2 mutations in endometrial cancer, melanoma, and gastric tumors (Tables 2 and 3).
Interestingly, somatic mutations in FGFR3 have been observed in benign skin conditions such as seborrheic keratosis and epidermal nevi (but not in adjacent normal skin) [54]. FGFR rearrangements are also observed in certain cancers and characterize certain myeloproliferative disorders (Table 2).
Importantly, there are several pharmacologic agents that have been or are being developed for inhibition of FGFR kinases. These include both highly selective inhibitors as well as multi-kinase inhibitors. Ponatinib, regorafenib, pazopanib, and lenvatinib are already FDA-approved for cancer, albeit not on the basis of their FGFR activity. Very few studies in cancer have been aimed at an FGFR biomarker-selected population, and several of the FGFR inhibitors have failed phase III studies in unselected patients. A multi-kinase inhibitor (nintedanib), which suppresses FGFR1–3, was also recently FDA-approved for idiopathic pulmonary fibrosis.
Whether or not FGFR inhibitors could also be used to moderate the phenotype of inherited disorders due to FGFR activation is an intriguing question. Of interest, Garcia et al. [[48](/article/10.1007/s10555-015-9579-8#ref-CR48 "Garcia, S., Dirat, B., Tognacci, T., Rochet, N., Mouska, X., Bonnafous, S., & Gouze, E. (2013). Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Science Translational Medicine, 5(203), 203ra–124. doi: 10.1126/scitranslmed.3006247
.")\] injected ia mouse model of achondroplasia with a soluble form of human FGFR3 (acting as a decoy receptor and preventing FGF from binding to mutant FGFR3), and effective maturation of growth plate chondrocytes was restored in the bones of treated mice. Of interest in this regard, individuals afflicted with the inherited disorders associated with FGFR aberrations, such as dwarfism, do not have an increased incidence of cancer, despite having mutations that are often identical to those somatic FGFR aberrations that characterize certain tumors. The secondary modulatory influences that mitigate the risk of cancer in these individuals are of interest. Whether or not treating them at an early age with FGFR inhibitors would increase the later risk of cancer if the inhibitors were withdrawn would need to be considered.In summary, perturbations in the FGF/FGFR machinery appear to underlie a variety of inherited syndromes, as well as benign and malignant disorders. The advent of potent FGFR inhibitors provides important new agents in the armamentarium against diseases caused by FGF/FGFR abnormalities.
References
- Hanahan, D., & Weinberg, R. A. (2000). The hallmarks of cancer. Cell, 100(1), 57–70.
Article CAS PubMed Google Scholar - Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of Cancer: The Next Generation. Cell, 144(5), 646–674. doi:10.1016/j.cell.2011.02.013.
Article CAS PubMed Google Scholar - Greenman, C., Stephens, P., Smith, R., Dalgliesh, G. L., Hunter, C., Bignell, G., & Stratton, M. R. (2007). Patterns of somatic mutation in human cancer genomes. Nature, 446(7132), 153–158.
Article PubMed Central CAS PubMed Google Scholar - Torkamani, A., & Schork, N. J. (2008). Prediction of cancer driver mutations in protein kinases. Cancer Research, 68(6), 1675–1682.
Article CAS PubMed Google Scholar - Muenke, M., Gripp, K. W., McDonald-McGinn, D. M., Gaudenz, K., Whitaker, L. A., Bartlett, S. P., & Wilkie, A. O. (1997). A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. American journal of human genetics, 60(3), 555–564.
PubMed Central CAS PubMed Google Scholar - Robin, N. H., Falk, M. J., & Haldeman-Englert, C. R. (1993). FGFR-Related Craniosynostosis Syndromes. In R. A. Pagon, M. P. Adam, H. H. Ardinger, T. D. Bird, C. R. Dolan, C.-T. Fong, … K. Stephens (Eds.), GeneReviews(®). Seattle (WA): University of Washington, Seattle. Retrieved from http://www.ncbi.nlm.nih.gov/books/NBK1455/.
- Anderson, J., Burns, H. D., Enriquez-Harris, P., Wilkie, A. O., & Heath, J. K. (1998). Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. Human molecular genetics, 7(9), 1475–1483.
Article CAS PubMed Google Scholar - He, L., Horton, W., & Hristova, K. (2010). Physical basis behind achondroplasia, the most common form of human dwarfism. The Journal of biological chemistry, 285(39), 30103–30114.
Article PubMed Central CAS PubMed Google Scholar - Rousseau, F., el Ghouzzi, V., Delezoide, A. L., Legeai-Mallet, L., Le Merrer, M., Munnich, A., & Bonaventure, J. (1996). Missense FGFR3 mutations create cysteine residues in thanatophoric dwarfism type I (TD1). Human molecular genetics, 5(4), 509–512.
Article CAS PubMed Google Scholar - Dodé, C., Teixeira, L., Levilliers, J., Fouveaut, C., Bouchard, P., Kottler, M.-L., & Hardelin, J.-P. (2006). Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS genetics, 2(10), e175.
Article PubMed Central PubMed CAS Google Scholar - OMIM hypogonatotrophic hypogonadism 2. (n.d.). Retrieved from http://www.omim.org/entry/147950.
- Dodé, C., Levilliers, J., Dupont, J.-M., De Paepe, A., Le Dû, N., Soussi-Yanicostas, N., & Hardelin, J.-P. (2003). Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nature Genetics, 33(4), 463–465. doi:10.1038/ng1122.
Article PubMed CAS Google Scholar - Thurman, R. D., Kathir, K. M., Rajalingam, D., & Kumar, T. K. S. (2012). Molecular basis for the Kallmann syndrome-linked fibroblast growth factor receptor mutation. Biochemical and Biophysical Research Communications, 425(3), 673–678. doi:10.1016/j.bbrc.2012.07.104.
Article PubMed Central CAS PubMed Google Scholar - Trarbach, E. B., Abreu, A. P., Silveira, L. F. G., Garmes, H. M., Baptista, M. T. M., Teles, M. G., & Latronico, A. C. (2010). Nonsense mutations in FGF8 gene causing different degrees of human gonadotropin-releasing deficiency. The Journal of Clinical Endocrinology and Metabolism, 95(7), 3491–3496. doi:10.1210/jc.2010-0176.
Article PubMed Central CAS PubMed Google Scholar - Turner, N., & Grose, R. (2010). Fibroblast growth factor signalling: from development to cancer. Nature Reviews Cancer, 10(2), 116–129.
Article CAS PubMed Google Scholar - Dienstmann, R., Rodon, J., Prat, A., Perez-Garcia, J., Adamo, B., Felip, E., & Tabernero, J. (2014). Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO, 25(3), 552–563. doi:10.1093/annonc/mdt419.
Article CAS Google Scholar - Baselga, J. (2006). Targeting tyrosine kinases in cancer: the second wave. Science, 312(5777), 1175–1178.
Article CAS PubMed Google Scholar - Yan, M., Parker, B. A., Schwab, R., & Kurzrock, R. (n.d.). HER2 aberrations in cancer: Implications for therapy. Cancer Treatment Reviews. doi:10.1016/j.ctrv.2014.02.008.
- Verstovsek, S., Mesa, R. A., Gotlib, J., Levy, R. S., Gupta, V., DiPersio, J. F., & Kantarjian, H. M. (2012). A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. New England Journal of Medicine, 366(9), 799–807. doi:10.1056/NEJMoa1110557.
Article CAS PubMed Google Scholar - Lee, E. B., Fleischmann, R., Hall, S., Wilkinson, B., Bradley, J. D., Gruben, D., & van Vollenhoven, R. F. (2014). Tofacitinib versus Methotrexate in Rheumatoid Arthritis. New England Journal of Medicine, 370(25), 2377–2386. doi:10.1056/NEJMoa1310476.
Article PubMed CAS Google Scholar - Hubbard, S. R., & Till, J. H. (2000). Protein tyrosine kinase structure and function. Annual review of biochemistry, 69, 373–398.
Article CAS PubMed Google Scholar - Kim, I., Moon, S.-O., Yu, K.-H., Kim, U.-H., & Koh, G. Y. (2001). A novel fibroblast growth factor receptor-5 preferentially expressed in the pancreas. Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression, 1518(1–2), 152–156.
Article CAS Google Scholar - Sleeman, M., Fraser, J., McDonald, M., Yuan, S., White, D., Grandison, P., & Murison, J. G. (2001). Identification of a new fibroblast growth factor receptor, FGFR5. Gene, 271(2), 171–182.
Article CAS PubMed Google Scholar - Klint, P., & Claesson-Welsh, L. (1999). Signal transduction by fibroblast growth factor receptors. Frontiers in bioscience : a journal and virtual library, 4, D165–77.
Article CAS Google Scholar - Jin, M., Du, X., & Chen, L. (2012). Cross-talk between FGF and other cytokine signalling pathways during endochondral bone development. Cell biology international, 36(8), 691–696.
Article CAS PubMed Google Scholar - Horton, W. A. (2006). MOLECULAR PATHOGENESIS OF ACHONDROPLASIA. Growth Genet Horm.
- Katoh, M. (2008). Cancer genomics and genetics of FGFR2 (Review). International journal of oncology, 33(2), 233–237.
CAS PubMed Google Scholar - Olsen, S. K., Garbi, M., Zampieri, N., Eliseenkova, A. V., Ornitz, D. M., Goldfarb, M., & Mohammadi, M. (2003). Fibroblast Growth Factor (FGF) Homologous Factors Share Structural but Not Functional Homology with FGFs. Journal of Biological \ldots.
- Itoh, N., & Ornitz, D. M. (2008). Functional evolutionary history of the mouse Fgf gene family. Developmental dynamics : an official publication of the American Association of Anatomists, 237(1), 18–27.
Article CAS Google Scholar - Potthoff, M. J., Kliewer, S. A., & Mangelsdorf, D. J. (2012). Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes & development, 26(4), 312–324.
Article CAS Google Scholar - Fukumoto, S. (2008). Actions and mode of actions of FGF19 subfamily members. Endocrine journal, 55(1), 23–31.
Article CAS PubMed Google Scholar - Guillemot, F., & Zimmer, C. (2011). From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron, 71(4), 574–588. doi:10.1016/j.neuron.2011.08.002.
Article CAS PubMed Google Scholar - Abuharbeid, S., Czubayko, F., & Aigner, A. (2006). The fibroblast growth factor-binding protein FGF-BP. The international journal of biochemistry & cell biology, 38(9), 1463–1468.
Article CAS Google Scholar - Zhang, X., Ibrahimi, O. A., Olsen, S. K., Umemori, H., Mohammadi, M., & Ornitz, D. M. (2006). Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. The Journal of biological chemistry, 281(23), 15694–15700.
Article PubMed Central CAS PubMed Google Scholar - Ornitz, D. M., Xu, J., Colvin, J. S., McEwen, D. G., MacArthur, C. A., Coulier, F., & Goldfarb, M. (1996). Receptor specificity of the fibroblast growth factor family. The Journal of biological chemistry, 271(25), 15292–15297.
Article CAS PubMed Google Scholar - Itoh, N. (2007). The Fgf families in humans, mice, and zebrafish: their evolutional processes and roles in development, metabolism, and disease. Biological & pharmaceutical bulletin, 30(10), 1819–1825.
Article CAS Google Scholar - Hehr, U., & Muenke, M. (1999). Craniosynostosis syndromes: from genes to premature fusion of skull bones. Molecular genetics and metabolism, 68(2), 139–151.
Article CAS PubMed Google Scholar - Wu, D. Q., Kan, M. K., Sato, G. H., Okamoto, T., & Sato, J. D. (1991). Characterization and molecular cloning of a putative binding protein for heparin-binding growth factors. The Journal of biological chemistry, 266(25), 16778–16785.
CAS PubMed Google Scholar - Böttcher, R. T., Pollet, N., Delius, H., & Niehrs, C. (2004). The transmembrane protein XFLRT3 forms a complex with FGF receptors and promotes FGF signalling. Nature cell biology, 6(1), 38–44.
Article PubMed CAS Google Scholar - Beenken, A., & Mohammadi, M. (2009). The FGF family: biology, pathophysiology and therapy. Nature reviews Drug discovery, 8(3), 235–253.
Article PubMed Central CAS PubMed Google Scholar - Hacohen, N., Kramer, S., Sutherland, D., Hiromi, Y., & Krasnow, M. A. (1998). sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell, 92(2), 253–263.
Article CAS PubMed Google Scholar - Cabrita, M. A., & Christofori, G. (2008). Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis, 11(1), 53–62.
Article CAS PubMed Google Scholar - Tsang, M., & Dawid, I. B. (2004). Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Science’s STKE : signal transduction knowledge environment, 2004(228), pe17.
- Steinberg, F., Zhuang, L., Beyeler, M., Kälin, R. E., Mullis, P. E., Brändli, A. W., & Trueb, B. (2010). The FGFRL1 receptor is shed from cell membranes, binds fibroblast growth factors (FGFs), and antagonizes FGF signaling in Xenopus embryos. The Journal of biological chemistry, 285(3), 2193–2202.
Article PubMed Central CAS PubMed Google Scholar - Trueb, B. (2011). Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cellular and molecular life sciences : CMLS, 68(6), 951–964.
Article CAS PubMed Google Scholar - Rieckmann, T., Zhuang, L., Flück, C. E., & Trueb, B. (2009). Characterization of the first FGFRL1 mutation identified in a craniosynostosis patient. Biochimica et biophysica acta, 1792(2), 112–121.
Article CAS PubMed Google Scholar - Schild, C., & Trueb, B. (2005). Aberrant expression of FGFRL1, a novel FGF receptor, in ovarian tumors. International journal of molecular medicine, 16(6), 1169–1173.
CAS PubMed Google Scholar - Garcia, S., Dirat, B., Tognacci, T., Rochet, N., Mouska, X., Bonnafous, S., & Gouze, E. (2013). Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Science Translational Medicine, 5(203), 203ra–124. doi:10.1126/scitranslmed.3006247.
Article CAS Google Scholar - Cohen, M. M. (2003). Neoplasms associated with alterations in fibroblast growth factor receptors. American journal of medical genetics Part A, 119A(2), 97–100.
Article PubMed Google Scholar - Andreou, A., Lamy, A., Layet, V., Cailliez, D., Gobet, F., Pfister, C., & Frebourg, T. (2006). Early-onset low-grade papillary carcinoma of the bladder associated with Apert syndrome and a germline FGFR2 mutation (Pro253Arg). American journal of medical genetics Part A, 140(20), 2245–2247.
Article PubMed CAS Google Scholar - Rouzier, C., Soler, C., Hofman, P., Brennetot, C., Bieth, E., & Pedeutour, F. (2008). Ovarian dysgerminoma and Apert syndrome. Pediatric blood & cancer, 50(3), 696–698.
Article Google Scholar - Barbosa, M., Almeida, M. do R., Reis-Lima, M., Pinto-Basto, J., & dos Santos, H. G. (2009). Muenke syndrome with osteochondroma. American journal of medical genetics. Part A, 149A(2), 260–261.
- Bourdeaut, F., Miquel, C., Di Rocco, F., Grison, C., Richer, W., Brugieres, L., & Collet, C. (2013). Germline mutations in FGF receptors and medulloblastomas. American journal of medical genetics Part A, 161A(2), 382–385.
Article PubMed CAS Google Scholar - Logié, A., Dunois-Lardé, C., Rosty, C., Levrel, O., Blanche, M., Ribeiro, A., & Radvanyi, F. (2005). Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Human molecular genetics, 14(9), 1153–1160.
Article PubMed CAS Google Scholar - Hafner, C., van Oers, J. M. M., Vogt, T., Landthaler, M., Stoehr, R., Blaszyk, H., & Hartmann, A. (2006). Mosaicism of activating FGFR3 mutations in human skin causes epidermal nevi. Journal of Clinical Investigation, 116(8), 2201–2207.
Article PubMed Central CAS PubMed Google Scholar - cbioportal. (n.d.). Retrieved from http://www.cbioportal.org/public-portal/index.do.
- Hattori, Y., Itoh, H., Uchino, S., Hosokawa, K., Ochiai, A., Ino, Y., & Terada, M. (1996). Immunohistochemical detection of K-sam protein in stomach cancer. Clinical cancer research : an official journal of the American Association for Cancer Research, 2(8), 1373–1381.
CAS Google Scholar - Gust, K. M., McConkey, D. J., Awrey, S., Hegarty, P. K., Qing, J., Bondaruk, J., & Black, P. C. (2013). Fibroblast growth factor receptor 3 is a rational therapeutic target in bladder cancer. Molecular Cancer Therapeutics, 12(7), 1245–1254.
Article PubMed Central CAS PubMed Google Scholar - Gartside, M. G., Chen, H., Ibrahimi, O. A., Byron, S. A., Curtis, A. V., Wellens, C. L., & Pollock, P. M. (2009). Loss-of-function fibroblast growth factor receptor-2 mutations in melanoma. Molecular cancer research : MCR, 7(1), 41–54.
Article PubMed Central CAS PubMed Google Scholar - Heist, R. S., Mino-Kenudson, M., Sequist, L. V., Tammireddy, S., Morrissey, L., Christiani, D. C., & Iafrate, A. J. (2012). FGFR1 Amplification in Squamous Cell Carcinoma of The Lung. Journal of Thoracic Oncology, 7(12), 1775–1780.
Article PubMed Central CAS PubMed Google Scholar - Weiss, J., Sos, M. L., Seidel, D., & Peifer, M. (2010). Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Science translational medicine, 2(62), 62ra93.
Article PubMed Central CAS PubMed Google Scholar - Albiges, L., Quidville, V., Valent, A., Mathieu, M. C., Drusch, F., Koscielny, S., … Andre, F. (2009). FGFR1 amplification and FGF gain in breast cancer. In San Antonio Breast Cancer Symposium. Institut Gustave Roussy, villejuif, France.
- Jackson, C. C., Medeiros, L. J., & Miranda, R. N. (2010). 8p11 myeloproliferative syndrome: a review. Human Pathology, 41(4), 461–476. doi:10.1016/j.humpath.2009.11.003.
Article CAS PubMed Google Scholar - Liu, J., Guzman, M. A., Pezanowski, D., Patel, D., Hauptman, J., Keisling, M., & de Chadarévian, J.-P. (2011). FOXO1-FGFR1 fusion and amplification in a solid variant of alveolar rhabdomyosarcoma. Modern Pathology: An Official Journal of the United States and Canadian Academy of Pathology, Inc, 24(10), 1327–1335. doi:10.1038/modpathol.2011.98.
Article CAS Google Scholar - Grand, E. K., Chase, A. J., Heath, C., Rahemtulla, A., & Cross, N. C. P. (2004). Targeting FGFR3 in multiple myeloma: inhibition of t(4;14)-positive cells by SU5402 and PD173074. Leukemia, 18(5), 962–966. doi:10.1038/sj.leu.2403347.
Article CAS PubMed Google Scholar - Yagasaki, F., Wakao, D., Yokoyama, Y., Uchida, Y., Murohashi, I., Kayano, H., & Bessho, M. (2001). Fusion of ETV6 to fibroblast growth factor receptor 3 in peripheral T-cell lymphoma with a t(4;12)(p16;p13) chromosomal translocation. Cancer Research, 61(23), 8371–8374.
CAS PubMed Google Scholar - Katoh, M., & Katoh, M. (2003). Recombination cluster around FGFR2-WDR11-HTPAPL locus on human chromosome 10q26. International Journal of Molecular Medicine, 11(5), 579–583.
CAS PubMed Google Scholar - Singh, D., Chan, J. M., Zoppoli, P., Niola, F., Sullivan, R., Castano, A., & Iavarone, A. (2012). Transforming Fusions of FGFR and TACC Genes in Human Glioblastoma. Science, 337(6099), 1231–1235.
Article PubMed Central CAS PubMed Google Scholar - Soria, J.-C., DeBraud, F., Bahleda, R., Adamo, B., Andre, F., Dientsmann, R., & Tabernero, J. (2014). Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO, 25(11), 2244–2251. doi:10.1093/annonc/mdu390.
Article Google Scholar - Nakanishi, Y., Akiyama, N., Tsukaguchi, T., Fujii, T., Sakata, K., Sase, H., & Ishii, N. (2014). The Fibroblast Growth Factor Receptor Genetic Status as a Potential Predictor of the Sensitivity to CH5183284/Debio 1347, a Novel Selective FGFR Inhibitor. Molecular Cancer Therapeutics, 13(11), 2547–2558. doi:10.1158/1535-7163.MCT-14-0248.
Article CAS PubMed Google Scholar - Bellovin, D. I., Palencia, S., Hestir, K., Lee, E., DeYoung, M. P., Brennan, T., & Baker, K. (2014). Abstract 5449: FP-1039/GSK3052230, an FGF ligand trap, enhances VEGF antagonist therapy in preclinical models of RCC and HCC. Cancer Research, 74(19 Supplement), 5449–5449. doi:10.1158/1538-7445.AM2014-5449.
Article Google Scholar - Sohl, C. D., Ryan, M. R., Luo, B., Frey, K. M., & Anderson, K. S. (2015). Illuminating the Molecular Mechanisms of Tyrosine Kinase Inhibitor Resistance for the FGFR1 Gatekeeper Mutation: The Achilles’ Heel of Targeted Therapy. ACS chemical biology. doi:10.1021/acschembio.5b00014.
PubMed Central PubMed Google Scholar - Chell, V., Balmanno, K., Little, A. S., Wilson, M., Andrews, S., Blockley, L., & Cook, S. J. (2013). Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene, 32(25), 3059–3070. doi:10.1038/onc.2012.319.
Article CAS PubMed Google Scholar - Byron, S. A., Chen, H., Wortmann, A., Loch, D., Gartside, M. G., Dehkhoda, F., & Pollock, P. M. (2013). The N550K/H mutations in FGFR2 confer differential resistance to PD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neoplasia (New York, N.Y.), 15(8), 975–988.
Article PubMed Central CAS Google Scholar - Crystal, A. S., Shaw, A. T., Sequist, L. V., Friboulet, L., Niederst, M. J., Lockerman, E. L., & Engelman, J. A. (2014). Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science, 346(6216), 1480–1486. doi:10.1126/science.1254721.
Article PubMed Central CAS PubMed Google Scholar - Harbinski, F., Craig, V. J., Sanghavi, S., Jeffery, D., Liu, L., Sheppard, K. A., & Tiedt, R. (2012). Rescue screens with secreted proteins reveal compensatory potential of receptor tyrosine kinases in driving cancer growth. Cancer Discovery, 2(10), 948–959. doi:10.1158/2159-8290.CD-12-0237.
Article CAS PubMed Google Scholar - Javidi-Sharifi, N., Traer, E., Martinez, J., Gupta, A., Taguchi, T., Dunlap, J., & Tyner, J. W. (2015). Crosstalk between KIT and FGFR3 Promotes Gastrointestinal Stromal Tumor Cell Growth and Drug Resistance. Cancer Research, 75(5), 880–891. doi:10.1158/0008-5472.CAN-14-0573.
Article CAS PubMed Google Scholar - Turner, N., Pearson, A., Sharpe, R., Lambros, M., Geyer, F., Lopez-Garcia, M. A., & Ashworth, A. (2010). FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer research, 70(5), 2085–2094. doi:10.1158/0008-5472.CAN-09-3746.
Article PubMed Central CAS PubMed Google Scholar - Schwaederlé, M., Daniels, G. A., Piccioni, D. E., Fanta, P. T., Schwab, R. B., Shimabukuro, K. A., … Kurzrock, R. (2014). Cyclin alterations in diverse cancers: Outcome and co-amplification network. Oncotarget.
- Wheler, J. J., Parker, B. A., Lee, J. J., Atkins, J. T., Janku, F., Tsimberidou, A. M., … Kurzrock, R. (2014). Unique molecular signatures as a hallmark of patients with metastatic breast cancer: Implications for current treatment paradigms. Oncotarget.
- Schwaederle, M., Parker, B. A., Schwab, R. B., Fanta, P. T., Boles, S. G., Daniels, G. A., & Kurzrock, R. (2014). Molecular tumor board: the University of California-San Diego Moores Cancer Center experience. The Oncologist, 19(6), 631–636. doi:10.1634/theoncologist.2013-0405.
Article PubMed Central PubMed Google Scholar - Tsimberidou, A.-M., Iskander, N. G., Hong, D. S., Wheler, J. J., Falchook, G. S., Fu, S., & Kurzrock, R. (2012). Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clinical cancer research: an official journal of the American Association for Cancer Research, 18(22), 6373–6383. doi:10.1158/1078-0432.CCR-12-1627.
Article CAS Google Scholar - omim.org. (n.d.). Retrieved from http://www.omim.org/.
- Cohen, M. M. (1993). Pfeiffer syndrome update, clinical subtypes, and guidelines for differential diagnosis. American journal of medical genetics, 45(3), 300–307.
Article PubMed Google Scholar - Vogels, A., & Fryns, J.-P. (2006). Pfeiffer syndrome. Orphanet journal of rare diseases, 1, 19.
Article PubMed Central PubMed Google Scholar - Ibrahimi, O. A., Zhang, F., Eliseenkova, A. V., Linhardt, R. J., & Mohammadi, M. (2004). Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through enhancement of FGF binding affinity. Human Molecular Genetics, 13(1), 69–78. doi:10.1093/hmg/ddh011.
Article CAS PubMed Google Scholar - Davies, H., Hunter, C., Smith, R., Stephens, P., Greenman, C., Bignell, G., … Futreal, P. A. (2005). Somatic Mutations of the Protein Kinase Gene Family in Human Lung Cancer.
- Ruhe, J. E., Streit, S., Hart, S., Wong, C.-H., Specht, K., Knyazev, P., … Ullrich, A. (2007). Genetic Alterations in the Tyrosine Kinase Transcriptome of Human Cancer Cell Lines. Cancer Research.
- White, K. E., Cabral, J. M., Davis, S. I., Fishburn, T., Evans, W. E., Ichikawa, S., & Econs, M. J. (2005). Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. American journal of human genetics, 76(2), 361–367.
Article PubMed Central CAS PubMed Google Scholar - Riminucci, M., Collins, M. T., Fedarko, N. S., Cherman, N., Corsi, A., White, K. E., & Gehron Robey, P. (2003). FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. Journal of Clinical Investigation, 112(5), 683–692.
Article PubMed Central CAS PubMed Google Scholar - Moloney, D. M., Slaney, S. F., Oldridge, M., Wall, S. A., Sahlin, P., Stenman, G., & Wilkie, A. O. (1996). Exclusive paternal origin of new mutations in Apert syndrome. Nature genetics, 13(1), 48–53.
Article CAS PubMed Google Scholar - Miraoui, H., Ringe, J., & Häupl, T. (2010). Increased EFG-and PDGFα-receptor signaling by mutant FGF-receptor 2 contributes to osteoblast dysfunction in Apert craniosynostosis. Human molecular \ldots.
- Pollock, P. M., Gartside, M. G., Dejeza, L. C., Powell, M. A., Mallon, M. A., Davies, H., & Goodfellow, P. J. (2007). Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene, 26(50), 7158–7162.
Article PubMed Central CAS PubMed Google Scholar - Byron, S. A., Gartside, M., Powell, M. A., Wellens, C. L., Gao, F., Mutch, D. G., & Pollock, P. M. (2012). FGFR2 point mutations in 466 endometrioid endometrial tumors: relationship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological features. PLoS ONE, 7(2), e30801.
Article PubMed Central CAS PubMed Google Scholar - Passos-Bueno, M. R., Wilcox, W. R., Jabs, E. W., Serti, A. L., Alonso, L. G., & Kitoh, H. (1999). Clinical spectrum of fibroblast growth factor receptor mutations. Human Mutation, 14(2), 115–125.
Article CAS PubMed Google Scholar - Reardon, W., Winter, R. M., Rutland, P., Pulleyn, L. J., Jones, B. M., & Malcolm, S. (1994). Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nature genetics, 8(1), 98–103.
Article CAS PubMed Google Scholar - Glaser, R. L., Jiang, W., Boyadjiev, S. A., Tran, A. K., Zachary, A. A., Van Maldergem, L., & Jabs, E. W. (2000). Paternal origin of FGFR2 mutations in sporadic cases of Crouzon syndrome and Pfeiffer syndrome. American journal of human genetics, 66(3), 768–777.
Article PubMed Central CAS PubMed Google Scholar - Jang, J. H., Shin, K. H., & Park, J. G. (2001). Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Research, 61(9), 3541–3543.
CAS PubMed Google Scholar - Chokdeemboon, C., Mahatumarat, C., Rojvachiranonda, N., Tongkobpetch, S., Suphapeetiporn, K., & Shotelersuk, V. (2013). FGFR1 and FGFR2 mutations in Pfeiffer syndrome. The Journal of craniofacial surgery, 24(1), 150–152.
Article PubMed Google Scholar - Cornejo-Roldan, L. R., Roessler, E., & Muenke, M. (1999). Analysis of the mutational spectrum of the FGFR2 gene in Pfeiffer syndrome - Springer. Human genetics.
- Chen, L., Adar, R., Yang, X., Monsonego, E. O., Li, C., Hauschka, P. V., & Deng, C. X. (1999). Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. Journal of Clinical Investigation, 104(11), 1517–1525.
Article PubMed Central CAS PubMed Google Scholar - Monsonego-Ornan, E., Adar, R., Feferman, T., Segev, O., & Yayon, A. (2000). The transmembrane mutation G380R in fibroblast growth factor receptor 3 uncouples ligand-mediated receptor activation from down-regulation. Molecular and cellular biology, 20(2), 516–522.
Article PubMed Central CAS PubMed Google Scholar - Henderson, J. E., Naski, M. C., Aarts, M. M., Wang, D., Cheng, L., Goltzman, D., & Ornitz, D. M. (2000). Expression of FGFR3 with the G380R Achondroplasia Mutation Inhibits Proliferation and Maturation of CFK2 Chondrocytic Cells. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research, 15(1), 155–165.
Article CAS Google Scholar - Knowles, M. A. (2008). Novel therapeutic targets in bladder cancer: mutation and expression of FGF receptors. Future oncology (London, England), 4(1), 71–83.
Article CAS Google Scholar - Ahmad, I., Iwata, T., & Leung, H. Y. (2012). Mechanisms of FGFR-mediated carcinogenesis. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 1823(4), 850–860.
Article CAS Google Scholar - Delezoide, A. L., Lasselin-Benoist, C., Legeai-Mallet, L., Brice, P., Senée, V., Yayon, A., & Bonaventure, J. (1997). Abnormal FGFR 3 expression in cartilage of thanatophoric dysplasia fetuses. Human molecular genetics, 6(11), 1899–1906.
Article CAS PubMed Google Scholar - Wilcox, W. R., Tavormina, P. L., Krakow, D., Kitoh, H., Lachman, R. S., Wasmuth, J. J., & Rimoin, D. L. (1998). Molecular, radiologic, and histopathologic correlations in thanatophoric dysplasia. American journal of medical genetics, 78(3), 274–281.
Article CAS PubMed Google Scholar - Brodie, S. G., Kitoh, H., Lachman, R. S., Nolasco, L. M., Mekikian, P. B., & Wilcox, W. R. (1999). Platyspondylic lethal skeletal dysplasia, San Diego type, is caused by FGFR3 mutations. American journal of medical genetics, 84(5), 476–480.
Article CAS PubMed Google Scholar - Di Martino, E., Tomlinson, D. C., & Knowles, M. A. (2012). A Decade of FGF Receptor Research in Bladder Cancer: Past, Present, and Future Challenges. Advances in Urology, 2012, e429213. doi:10.1155/2012/429213.
Article Google Scholar - Salgia, R. (2014). Fibroblast growth factor signaling and inhibition in non-small cell lung cancer and their role in squamous cell tumors. Cancer Medicine, 3(3), 681–692. doi:10.1002/cam4.238.
Article PubMed Central CAS PubMed Google Scholar - Liao, R. G., Jung, J., Tchaicha, J., Wilkerson, M. D., Sivachenko, A., Beauchamp, E. M., & Hammerman, P. S. (2013). Inhibitor-Sensitive FGFR2 and FGFR3 Mutations in Lung Squamous Cell Carcinoma. Cancer Research, 73(16), 5195–5205. doi:10.1158/0008-5472.CAN-12-3950.
Article PubMed Central CAS PubMed Google Scholar - Webster, M. K., D’Avis, P. Y., Robertson, S. C., & Donoghue, D. J. (1996). Profound ligand-independent kinase activation of fibroblast growth factor receptor 3 by the activation loop mutation responsible for a lethal skeletal dysplasia, thanatophoric dysplasia type II. Molecular and cellular biology, 16(8), 4081–4087.
Article PubMed Central CAS PubMed Google Scholar - Lievens, P. M.-J., & Liboi, E. (2003). The thanatophoric dysplasia type II mutation hampers complete maturation of fibroblast growth factor receptor 3 (FGFR3), which activates signal transducer and activator of transcription 1 (STAT1) from the endoplasmic reticulum. The Journal of biological chemistry, 278(19), 17344–17349.
Article CAS PubMed Google Scholar - Otsuka, M., Mizuki, M., Fujita, J., Kang, S., & Kanakura, Y. (2011, January). Constitutively active FGFR3 with Lys650Glu mutation enhances bortezomib sensitivity in plasma cell malignancy. Anticancer Research.
- Walker, B. A., Murdoch, J. L., McKusick, V. A., Langer, L. O., & Beals, R. K. (1971). Hypochondroplasia. American journal of diseases of children (1960), 122(2), 95–104.
CAS Google Scholar - Bellus, G. A., McIntosh, I., Smith, E. A., Aylsworth, A. S., Kaitila, I., Horton, W. A., & Francomano, C. A. (1995). A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nature genetics, 10(3), 357–359.
Article CAS PubMed Google Scholar - Bellus, G. A., Garber, A. T., Bryke, C. R., Weaver, C. A., Speiser, P. W., Webster, M. K., & Spector, E. B. (2000). FGFR3 Mutations K650N and K650Q Cause Hypochondroplasia. Genetics in Medicine, 2(1), 76–76.
Article Google Scholar - Heuertz, S., Le Merrer, M., Zabel, B., Wright, M., Legeai-Mallet, L., Cormier-Daire, V., & Bonaventure, J. (2006). Novel FGFR3 mutations creating cysteine residues in the extracellular domain of the receptor cause achondroplasia or severe forms of hypochondroplasia. European Journal of Human Genetics, 14(12), 1240–1247.
Article CAS PubMed Google Scholar - Abdel-Salam, G. M. H., Flores-Sarnat, L., El-Ruby, M. O., Parboosingh, J., Bridge, P., Eid, M. M., & Temtamy, S. A. (2011). Muenke syndrome with pigmentary disorder and probable hemimegalencephaly: An expansion of the phenotype. American journal of medical genetics Part A, 155A(1), 207–214.
Article PubMed Google Scholar - Cancer Genome Atlas Network. (2012). Comprehensive molecular portraits of human breast tumours. Nature, 490(7418), 61–70. doi:10.1038/nature11412.
Article CAS Google Scholar - Shiang, C. Y., Qi, Y., Wang, B., Lazar, V., Wang, J., Fraser Symmans, W., & Pusztai, L. (2010). Amplification of fibroblast growth factor receptor-1 in breast cancer and the effects of brivanib alaninate. Breast Cancer Research and Treatment, 123(3), 747–755.
Article CAS PubMed Google Scholar - Brunello, E., Brunelli, M., Bogina, G., Caliò, A., Manfrin, E., Nottegar, A., & Bonetti, F. (2012). FGFR-1 amplification in metastatic lymph-nodal and haematogenous lobular breast carcinoma. Journal of experimental & clinical cancer research : CR, 31, 103.
Article PubMed Central CAS Google Scholar - Elbauomy Elsheikh, S., Green, A. R., Lambros, M. B. K., Turner, N. C., Grainge, M. J., Powe, D., & Reis-Filho, J. S. (2007). FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Research, 9(2), R23.
Article PubMed Central PubMed CAS Google Scholar - The Cancer Genome Atlas Research Network. (2014). Comprehensive molecular characterization of urothelial bladder carcinoma. Nature, 507(7492), 315–322. doi:10.1038/nature12965.
Article PubMed Central CAS Google Scholar - Freier, K., Schwaenen, C., Sticht, C., Flechtenmacher, C., Mühling, J., Hofele, C., & Joos, S. (2007). Recurrent FGFR1 amplification and high FGFR1 protein expression in oral squamous cell carcinoma (OSCC). Oral oncology, 43(1), 60–66.
Article CAS PubMed Google Scholar - Goke, F., Franzen, A., Roopika, M., Schroeck, A., Kirsten, R., Boehm, D., & Perner, S. (2013). In 25th European Congress of Pathology. Portugal: Lisbon.
Google Scholar - The Cancer Genome Atlas Research Network. (2014). Comprehensive molecular characterization of gastric adenocarcinoma. Nature, 513(7517), 202–209. doi:10.1038/nature13480.
Article CAS Google Scholar - Xie, L., Su, X., Zhang, L., Yin, X., Tang, L., Zhang, X., … Ji, Q. (2013). FGFR2 gene amplification in gastric cancer predicts sensitivity to the selective FGFR inhibitor AZD4547. Clinical cancer research : an official journal of the American Association for Cancer Research.
- Matsumoto, K., Arao, T., Hamaguchi, T., Shimada, Y., Kato, K., Oda, I., & Yamada, Y. (2012). FGFR2 gene amplification and clinicopathological features in gastric cancer. British Journal of Cancer, 106(4), 727–732.
Article PubMed Central CAS PubMed Google Scholar - Cancer Genome Atlas Research Network. (2011). Integrated genomic analyses of ovarian carcinoma. Nature, 474(7353), 609–615.
Article CAS Google Scholar - Barretina, J., Taylor, B. S., Banerji, S., Ramos, A. H., Lagos-Quintana, M., Decarolis, P. L., & Singer, S. (2010). Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nature genetics, 42(8), 715–721.
Article PubMed Central CAS PubMed Google Scholar - Hodis, E., Watson, I. R., Kryukov, G. V., Arold, S. T., Imielinski, M., Theurillat, J.-P., & Chin, L. (2012). A Landscape of Driver Mutations in Melanoma. Cell, 150(2), 251–263. doi:10.1016/j.cell.2012.06.024.
Article PubMed Central CAS PubMed Google Scholar - Cancer Genome Atlas Research Network, Kandoth, C., Schultz, N., Cherniack, A. D., Akbani, R., Liu, Y., & Levine, D. A. (2013). Integrated genomic characterization of endometrial carcinoma. Nature, 497(7447), 67–73.
Article CAS Google Scholar - Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. (2002). , Published online: 03 December 2002; | doi: 10.1038/sj.ejhg.5200883 , 10(12). doi:10.1038/sj.ejhg.5200883.
- Williams, S. V., Hurst, C. D., & Knowles, M. A. (2013). Oncogenic FGFR3 gene fusions in bladder cancer. Human molecular genetics, 22(4), 795–803.
Article PubMed Central CAS PubMed Google Scholar - Rosty, C., Aubriot, M.-H., Cappellen, D., Bourdin, J., Cartier, I., Thiery, J. P., & Radvanyi, F. (2005). Clinical and biological characteristics of cervical neoplasias with FGFR3 mutation. Molecular Cancer, 4, 15. doi:10.1186/1476-4598-4-15.
Article PubMed Central PubMed CAS Google Scholar - Taylor, J. G., Cheuk, A. T., Tsang, P. S., Chung, J.-Y., Song, Y. K., Desai, K., & Khan, J. (2009). Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. The Journal of Clinical Investigation, 119(11), 3395–3407. doi:10.1172/JCI39703.
CAS PubMed Google Scholar - Turkington, R. C., Longley, D. B., Allen, W. L., Stevenson, L., McLaughlin, K., Dunne, P. D., & Johnston, P. G. (2014). Fibroblast growth factor receptor 4 (FGFR4): a targetable regulator of drug resistance in colorectal cancer. Cell Death & Disease, 5(2), e1046. doi:10.1038/cddis.2014.10.
Article CAS Google Scholar - Persson, F., Winnes, M., Andrén, Y., Wedell, B., Dahlenfors, R., Asp, J., & Stenman, G. (2008). High-resolution array CGH analysis of salivary gland tumors reveals fusion and amplification of the FGFR1 and PLAG1 genes in ring chromosomes. Oncogene, 27(21), 3072–3080. doi:10.1038/sj.onc.1210961.
Article CAS PubMed Google Scholar - Arai, Y., Totoki, Y., Hosoda, F., Shirota, T., Hama, N., Nakamura, H., & Shibata, T. (2014). Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology, 59(4), 1427–1434. doi:10.1002/hep.26890.
Article CAS PubMed Google Scholar - Wu, Y.-M., Su, F., Kalyana-Sundaram, S., Khazanov, N., Ateeq, B., Cao, X., … Chinnaiyan, A. M. (2013). Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer discovery.
- Cancer Genome Atlas Research Network. (2012). Comprehensive genomic characterization of squamous cell lung cancers. Nature, 489(7417), 519–525. doi:10.1038/nature11404.
Article CAS Google Scholar - Shah, S. P., Roth, A., Goya, R., Oloumi, A., Ha, G., Zhao, Y., & Aparicio, S. (2012). The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature, 486(7403), 395–399.
CAS PubMed Google Scholar - Banerji, S., Cibulskis, K., Rangel-Escareno, C., Brown, K. K., Carter, S. L., Frederick, A. M., & Meyerson, M. (2012). Sequence analysis of mutations and translocations across breast cancer subtypes. Nature, 486(7403), 405–409.
Article PubMed Central CAS PubMed Google Scholar - Stephens, P. J., Tarpey, P. S., Davies, H., Van Loo, P., Greenman, C., Wedge, D. C., & Stratton, M. R. (2012). The landscape of cancer genes and mutational processes in breast cancer. Nature, 486(7403), 400–404.
PubMed Central CAS PubMed Google Scholar - Gaykalova, D. A., Mambo, E., Choudhary, A., Houghton, J., Buddavarapu, K., Sanford, T., & Sun, W. (2014). Novel insight into mutational landscape of head and neck squamous cell carcinoma. PloS One, 9(3), e93102. doi:10.1371/journal.pone.0093102.
Article PubMed Central PubMed CAS Google Scholar - Mountzios, G., Rampias, T., & Psyrri, A. (2014). The mutational spectrum of squamous-cell carcinoma of the head and neck: targetable genetic events and clinical impact. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. doi:10.1093/annonc/mdu143.
Google Scholar - The Cancer Genome Atlas Research Network. (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature, 511(7511), 543–550. doi:10.1038/nature13385.
Article PubMed Central CAS Google Scholar - Imielinski, M., Berger, A. H., Hammerman, P. S., Hernandez, B., Pugh, T. J., Hodis, E., & Meyerson, M. (2012). Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell, 150(6), 1107–1120.
Article PubMed Central CAS PubMed Google Scholar - Ding, L., Getz, G., Wheeler, D. A., Mardis, E. R., McLellan, M. D., Cibulskis, K., & Wilson, R. K. (2008). Somatic mutations affect key pathways in lung adenocarcinoma. Nature, 455(7216), 1069–1075.
Article PubMed Central CAS PubMed Google Scholar - Taylor, B. S., Schultz, N., Hieronymus, H., Gopalan, A., Xiao, Y., Carver, B. S., & Gerald, W. L. (2010). Integrative genomic profiling of human prostate cancer. Cancer Cell, 18(1), 11–22.
Article PubMed Central CAS PubMed Google Scholar - The Cancer Genome Atlas Research Network. (2013). Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature, 499(7456), 43–49. doi:10.1038/nature12222.
Article PubMed Central CAS Google Scholar - ancer Genome Atlas Network. (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature, 487(7407), 330–337. doi:10.1038/nature11252.
Article CAS Google Scholar - McLendon, R., Friedman, A., Bigner, D., Van Meir, E. G., Brat, D. J., Mastrogianakis, M. G., & Thomson, E. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature, 455(7216), 1061–1068.
Article CAS Google Scholar - Ho, A. S., Kannan, K., Roy, D. M., Morris, L. G. T., Ganly, I., Katabi, N., & Chan, T. A. (2013). The mutational landscape of adenoid cystic carcinoma. Nature genetics, 45(7), 791–798.
Article CAS PubMed Google Scholar - Brat, D. J. (2014). Integrative Genomic Characterization of Lower Grade Gliomas. Neuro-Oncology, 16, iii3. doi:10.1093/neuonc/nou206.11.
Article Google Scholar - Cancer Genome Atlas Research Network. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine, 368(22), 2059–2074.
Article CAS Google Scholar - Seshagiri, S., Stawiski, E. W., Durinck, S., Modrusan, Z., Storm, E. E., Conboy, C. B., & de Sauvage, F. J. (2012). Recurrent R-spondin fusions in colon cancer. Nature, 488(7413), 660–664.
Article PubMed Central CAS PubMed Google Scholar - clinicaltrials.gov. (n.d.). Retrieved from http://clinicaltrials.gov/ct2/home.
- Smyth, E. C., Turner, N. C., Popat, S., Morgan, S., Owen, K., Gillbanks, A., & Cunningham, D. (2013). FGFR: Proof-of-concept study of AZD4547 in patients with FGFR1 or FGFR2 amplified tumours. ASCO Meeting Abstracts, 31(15_suppl), TPS2626.
Google Scholar - Zhang, J., Zhang, L., Su, X., Li, M., Xie, L., Malchers, F., & Gavine, P. R. (2012). Translating the therapeutic potential of AZD4547 in FGFR1-amplified non-small cell lung cancer through the use of patient-derived tumor xenograft models. Clinical cancer research : an official journal of the American Association for Cancer Research, 18(24), 6658–6667.
Article CAS Google Scholar - Johnson, P. J., Qin, S., Park, J.-W., Poon, R. T. P., Raoul, J.-L., Philip, P. A., & Cheng, A.-L. (2013). Brivanib Versus Sorafenib As First-Line Therapy in Patients With Unresectable, Advanced Hepatocellular Carcinoma: Results From the Randomized Phase III BRISK-FL Study. Journal of Clinical Oncology, JCO, 2012(48), 4410. doi:10.1200/JCO.2012.48.4410.
Google Scholar - Huynh, H., Ngo, V. C., Fargnoli, J., Ayers, M., Soo, K. C., Koong, H. N., & Tran, E. (2008). Brivanib Alaninate, a Dual Inhibitor of Vascular Endothelial Growth Factor Receptor and Fibroblast Growth Factor Receptor Tyrosine Kinases, Induces Growth Inhibition in Mouse Models of Human Hepatocellular Carcinoma. Clinical Cancer Research, 14(19), 6146–6153.
Article CAS PubMed Google Scholar - Siu, L. L., Shapiro, J. D., Jonker, D. J., Karapetis, C. S., Zalcberg, J. R., Simes, J., … NCIC Clinical Trials Group and AGITG. (2012). Phase III randomized trial of cetuximab (CET) plus either brivanib alaninate (BRIV) or placebo in patients (pts) with metastatic (MET) chemotherapy refractory K-RAS wild-type (WT) colorectal carcinoma (CRC): The NCIC Clinical Trials Group and AGITG CO.20 trial. ASCO Meeting Abstracts, 30(4_suppl), 386.
- Meulenbeld, H. J., Bleuse, J. P., Vinci, E. M., Raymond, E., Vitali, G., Santoro, A., & de Wit, R. (2013). Randomized phase II study of danusertib in patients with metastatic castration-resistant prostate cancer after docetaxel failure. BJU international, 111(1), 44–52. doi:10.1111/j.1464-410X.2012.11404.x.
Article CAS PubMed Google Scholar - Cohen, R. B., Jones, S. F., Aggarwal, C., von Mehren, M., Cheng, J., Spigel, D. R., & Burris, H. A. (2009). A phase I dose-escalation study of danusertib (PHA-739358) administered as a 24-hour infusion with and without granulocyte colony-stimulating factor in a 14-day cycle in patients with advanced solid tumors. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 15(21), 6694–6701. doi:10.1158/1078-0432.CCR-09-1445.
Article CAS Google Scholar - Motzer, R. J., Porta, C., Vogelzang, N. J., Sternberg, C. N., Szczylik, C., Zolnierek, J., & Escudier, B. (2014). Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: an open-label, randomised phase 3 trial. The Lancet. Oncology, 15(3), 286–296. doi:10.1016/S1470-2045(14)70030-0.
Article CAS PubMed Google Scholar - Motzer, R. J., Porta, C., Bjarnason, G. A., Szcylik, C., Rha, S. Y., Esteban, E., … Escudier, B. J. (2012). Phase III trial of dovitinib (TKI258) versus sorafenib in patients with metastatic renal cell carcinoma after failure of anti-angiogenic (VEGF-targeted and mTOR inhibitor) therapies. ASCO Meeting Abstracts, 30(15_suppl), TPS4683.
- Angevin, E., Lin, C., Pande, A. U., & Lopez, J. A. (2010). A phase I/II study of dovitinib (TKI258), a FGFR and VEGFR inhibitor, in patients (pts) with advanced or metastatic renal cell cancer: phase I results. ASCO Meeting \ldots.
- Dienstmann, R., Bahleda, R., Adamo, B., Rodon, J., Varga, A., Gazzah, A., & Soria, J.-C. (2014). Abstract CT325: First in human study of JNJ-42756493, a potent pan fibroblast growth factor receptor (FGFR) inhibitor in patients with advanced solid tumors. Cancer Research, 74(19 Supplement), CT325–CT325. doi:10.1158/1538-7445.AM2014-CT325.
Article Google Scholar - Schlumberger, M., Tahara, M., Wirth, L. J., Robinson, B., Brose, M. S., Elisei, R., … Sherman, S. I. (2014). A phase 3, multicenter, double-blind, placebo-controlled trial of lenvatinib (E7080) in patients with 131I-refractory differentiated thyroid cancer (SELECT) [abstract]. In Journal of clinical oncology : official journal of the American Society of Clinical Oncology (p. LBA6008).
- Tohyama, O., Matsui, J., Kodama, K., Hata-Sugi, N., Kimura, T., Okamoto, K., & Funahashi, Y. (2014). Antitumor Activity of Lenvatinib (E7080): An Angiogenesis Inhibitor That Targets Multiple Receptor Tyrosine Kinases in Preclinical Human Thyroid Cancer Models. Journal of Thyroid Research, 2014, e638747. doi:10.1155/2014/638747.
Article CAS Google Scholar - Andre, F., Daly, F., Azim, H. A., Agrapart, V., Goulioti, T., Pinto, A. C. D. C. P., … Cortes, J. (2014). FINESSE: An open, three-cohort, phase II trial testing oral administration of lucitanib in patients with FGFR1-amplified or nonamplified estrogen receptor-positive metastatic breast cancer. Journal of Clinical Oncology, 32:5s(suppl; abstr TPS1134). Retrieved from http://meetinglibrary.asco.org/content/126911-144.
- Bello, E., Taraboletti, G., Colella, G., Zucchetti, M., Forestieri, D., Licandro, S. A., & Damia, G. (2013). The Tyrosine Kinase Inhibitor E-3810 Combined with Paclitaxel Inhibits the Growth of Advanced-Stage Triple-Negative Breast Cancer Xenografts. Molecular Cancer Therapeutics, 12(2), 131–140.
Article CAS PubMed Google Scholar - Bello, E., Colella, G., Scarlato, V., Oliva, P., Berndt, A., Valbusa, G., & Camboni, G. (2011). E-3810 is a potent dual inhibitor of VEGFR and FGFR that exerts antitumor activity in multiple preclinical models. Cancer Research, 71(4), 1396–1405.
Article CAS PubMed Google Scholar - Carter, E. P., Fearon, A. E., & Grose, R. P. (n.d.). Careless talk costs lives: fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends in Cell Biology. doi:10.1016/j.tcb.2014.11.003.
- Pan, B.-S., Chan, G. K. Y., Chenard, M., Chi, A., Davis, L. J., Deshmukh, S. V., & Dinsmore, C. J. (2010). MK-2461, a novel multitargeted kinase inhibitor, preferentially inhibits the activated c-Met receptor. Cancer Research, 70(4), 1524–1533.
Article CAS PubMed Google Scholar - Hanna, N. H., Kaiser, R., Sullivan, R. N., Aren, O. R., Ahn, M.-J., Tiangco, B., … Kim, J. H. (2013). Lume-lung 2: A multicenter, randomized, double-blind, phase III study of nintedanib plus pemetrexed versus placebo plus pemetrexed in patients with advanced nonsquamous non-small cell lung cancer (NSCLC) after failure of first-line chemotherapy. ASCO Meeting Abstracts, 31(15_suppl), 8034.
- Richeldi, L., du Bois, R. M., Raghu, G., Azuma, A., Brown, K. K., Costabel, U., & Collard, H. R. (2014). Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. New England Journal of Medicine, 370(22), 2071–2082. doi:10.1056/NEJMoa1402584.
Article PubMed CAS Google Scholar - Kristensen, G., Harter, P., Trédan, O., Sailer, M. O., Bamias, A., Colombo, N., … Bois, A. D. (2014). Independent review of AGO-OVAR 12, a GCIG/ENGOT-Intergroup phase III trial of nintedanib (N) in first-line therapy for ovarian cancer (OC). Journal of Clinical Oncology, 32:5s(suppl; abstr 5556^). Retrieved from http://meetinglibrary.asco.org/content/130341-144.
- Guagnano, V., Furet, P., Spanka, C., Bordas, V., Le Douget, M., Stamm, C., & Graus Porta, D. (2011). Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. Journal of Medicinal Chemistry, 54(20), 7066–7083. doi:10.1021/jm2006222.
Article CAS PubMed Google Scholar - Guagnano, V., Kauffmann, A., Wöhrle, S., Stamm, C., Ito, M., Barys, L., & Graus-Porta, D. (2012). FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discovery, 2(12), 1118–1133. doi:10.1158/2159-8290.CD-12-0210.
Article CAS PubMed Google Scholar - Konecny, G. E., Kolarova, T., O’Brien, N. A., Winterhoff, B., Yang, G., Qi, J., … Slamon, D. J. (2013). Activity of the Fibroblast Growth Factor Receptor Inhibitors Dovitinib (TKI258) and NVP-BGJ398 in Human Endometrial Cancer Cells. Molecular Cancer Therapeutics.
- Motzer, R. J., Hutson, T. E., McCann, L., Deen, K., & Choueiri, T. K. (2014). Overall survival in renal-cell carcinoma with pazopanib versus sunitinib. The New England Journal of Medicine, 370(18), 1769–1770. doi:10.1056/NEJMc1400731.
Article CAS PubMed Google Scholar - Kasper, B., Sleijfer, S., Litière, S., Marreaud, S., Verweij, J., Hodge, R. A., & van der Graaf, W. T. A. (2014). Long-term responders and survivors on pazopanib for advanced soft tissue sarcomas: subanalysis of two European Organisation for Research and Treatment of Cancer (EORTC) clinical trials 62043 and 62072. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO, 25(3), 719–724. doi:10.1093/annonc/mdt586.
Article CAS Google Scholar - O’Hare, T., Shakespeare, W. C., Zhu, X., Eide, C. A., Rivera, V. M., Wang, F., & Clackson, T. (2009). AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell, 16(5), 401–412.
Article PubMed Central PubMed CAS Google Scholar - Cortes, J. E., Kim, D.-W., Pinilla-Ibarz, J., le Coutre, P., Paquette, R., Chuah, C., & Investigators, P. A. C. E. (2013). A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. The New England Journal of Medicine, 369(19), 1783–1796. doi:10.1056/NEJMoa1306494.
Article CAS PubMed Google Scholar - Demetri, G. D., Reichardt, P., Kang, Y.-K., Blay, J.-Y., Rutkowski, P., Gelderblom, H., & GRID study investigators. (2013). Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet, 381(9863), 295–302. doi:10.1016/S0140-6736(12)61857-1.
Article CAS PubMed Google Scholar - Grothey, A., Van Cutsem, E., Sobrero, A., Siena, S., Falcone, A., Ychou, M., & CORRECT Study Group. (2013). Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet, 381(9863), 303–312. doi:10.1016/S0140-6736(12)61900-X.
Article CAS PubMed Google Scholar - Ochiiwa, H., Fujita, H., Itoh, K., Sootome, H., Hashimoto, A., Fujioka, Y., & Utsugi, T. (2013). Abstract A270: TAS-120, a highly potent and selective irreversible FGFR inhibitor, is effective in tumors harboring various FGFR gene abnormalities. Molecular Cancer Therapeutics, 12(11 Supplement), A270–A270. doi:10.1158/1535-7163.TARG-13-A270.
Article Google Scholar - Powers, C. J., McLeskey, S. W., & Wellstein, A. (2000). Fibroblast growth factors, their receptors and signaling. Endocrine-Related Cancer, 7(3), 165–197.
Article CAS PubMed Google Scholar - Itoh, N., & Ornitz, D. M. (2004). Evolution of the Fgf and Fgfr gene families. Trends in Genetics, 20(11), 563–569. doi:10.1016/j.tig.2004.08.007.
Article CAS PubMed Google Scholar