Regulation of DNA double-strand break repair pathway choice (original) (raw)
Introduction
DNA double-strand breaks (DSBs) pose a serious threat to cell viability and genome stability. DSBs are generated naturally when replication forks encounter blocking lesions such as those produced by metabolic byproducts of cellular respiration (reactive oxygen species (ROS)) leading to fork collapse 1; during programmed genome rearrangements induced by nucleases, including yeast mating-type switching 2, V(D)J recombination 3, class-switch recombination 4, and meiosis 5; and from physical stress when dicentric or catenated chromosomes are pulled to opposite poles during mitosis 6, 7. DSBs are also produced when cells are exposed to DNA damaging agents including ionizing radiation (IR), which creates DSBs directly and indirectly via production of ROS 8; chemical agents and UV light that create replication blocking lesions (alkyl adducts, pyrimidine dimers, and crosslinks) 9, 10; and cancer chemotherapeutics that poison topoisomerase I, which produces replication-blocking lesions, or topoisomerase II, which traps the enzyme-DNA complex after DSB induction and can potentially produce DSBs during any phase of the cell cycle 11. The failure to repair DSBs, or misrepair, can result in cell death or large-scale chromosome changes including deletions, translocations, and chromosome fusions that enhance genome instability and are hallmarks of cancer cells. Cells have evolved groups of proteins that function in signaling networks that sense DSBs or other DNA damage, arrest the cell cycle, and activate DNA repair pathways. These cellular responses can occur at various stages of the cell cycle and are collectively called DNA damage checkpoints, but when cells suffer too much damage overlapping signaling pathways can trigger apoptosis to prevent propagation of cells with highly unstable genomes 12.
Eukaryotic cells repair DSBs primarily by two mechanisms: nonhomologous end-joining (NHEJ) and homologous recombination (HR). Frank DSBs, such as those produced by nucleases and IR, can be repaired by either pathway. DSBs produced by replication fork collapse are repaired primarily (or perhaps exclusively) by HR 1, 13. Note that fork collapse produces a one-ended DSB, better described as a “double-strand end” (DSE). Because a DSE at a collapsed fork has no second end with which to rejoin, it is difficult to imagine how NHEJ can contribute to the repair of collapsed replication forks (Figure 1A). However, this does not rule out indirect roles for NHEJ proteins in replication fork restart (see below). Here we review factors that regulate DSB repair pathway choice, and therefore the discussion is focused primarily on the repair of frank DSBs (Figure 1B).
Figure 1
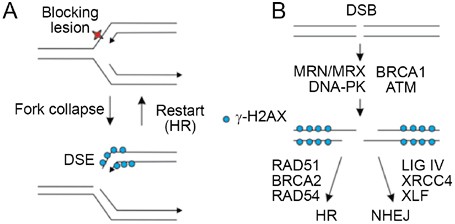
Differences between DSEs and DSBs. (A) Replication forks stall and may collapse when they encounter a blocking lesion or a nick, producing a DSE which induces H2AX phosphorylation adjacent to the break. Fork restart typically involves HR proteins. (B) A frank DSB may be repaired by NHEJ or HR. There is rapid phosphorylation of H2AX adjacent to DSBs, but in the immediate vicinity of the broken ends nucleosome eviction (and perhaps other processes such as histone exchange) results in reduced levels of γ-H2AX. Repair pathway choice may be controlled by the early acting proteins that influence both repair pathways. Once the commitment is made to a repair pathway, pathway-specific proteins drive the reaction toward HR or NHEJ products.
NHEJ and HR both contribute to genome stability and both pose risks of large- and small-scale genome rearrangement
NHEJ and HR pathways are often described as “error-prone” and “error-free” respectively, but this is an oversimplification. “Clean” DSBs with complementary overhangs, 5′ phosphates and 3′ hydroxyl groups, such as those produced by nucleases, can be precisely repaired by NHEJ. In yeast and mammalian cells, 25-50% of nuclease DSBs are repaired by precise NHEJ 14, 15; note that these are minimum estimates because these measurements do not account for multiple cycles of cleavage and precise repair. When ends cannot be precisely rejoined, NHEJ typically involves alignment of one or a few complementary bases (“microhomology”) to direct repair, leading to small deletions and sometimes small insertions. In mammalian cells NHEJ proceeds in a stepwise manner beginning with limited end-processing by the MRE11/RAD50/NBS1 (MRN) complex and perhaps other factors, end-binding by Ku comprising the Ku70 and Ku80 subunits, and recruitment of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), forming the trimeric DNA-PK holoenzyme. Once bound to broken ends, DNA-PK is activated and it phosphorylates itself and other targets including RPA, WRN, and Artemis; in cells lacking ATM, DNA-PK can also phosphorylate histone H2AX, termed γ-H2AX 16, 17, 18, 19, 20, 21, 22, 23, 24. In the final step, DNA ligase IV, with its binding partners XRCC4 and XLF (also called Cernunnos), seals the break. The nuclease Artemis helps repair a subset of IR-induced DSBs by NHEJ, and is important for opening hairpins formed during V(D)J recombination 25, 26. Terminal deoxynucleotidyl transferase (TdT) and two members of the polymerase X family (pol μ and pol λ) can modify NHEJ outcomes by non-templated addition of nucleotides to ends or by extending a 3′ single-stranded DNA (ssDNA) tail that can transiently pair via microhomology on the other broken end 27. The breast cancer tumor suppressor protein BRCA1 has a role in NHEJ that may involve modulation of MRE11 28, or chromatin remodeling via the Fanconi anemia ubiquitylation pathway 29. An alternative Ligase III-mediated NHEJ pathway is promoted by PARP-1 and is more error-prone than classical NHEJ 30. The yeast NHEJ machinery includes homologs of all of these proteins except DNA-PKcs, Artemis, and BRCA1. Further details about NHEJ protein biochemistry and repair mechanisms can be found in recent reviews 31, 32, 33, 34 and in articles in this volume by Weterings and Chen, and Lieber et al.
HR is considered a more accurate mechanism for DSB repair because broken ends use homologous sequences elsewhere in the genome (sister chromatids, homologous chromosomes, or repeated regions on the same or different chromosomes) to prime repair synthesis. If the repair template is perfectly homologous, repair can be 100% accurate, although even in this case there is evidence from yeast that the repair polymerase is more error-prone than replicative polymerases, and point mutations arise at increased frequencies adjacent to DSB repair sites 35. With the exception of sister chromatids, repair templates are often not perfectly homologous, and in these cases HR results in loss of heterozygosity, with information transferred non-reciprocally from the unbroken (donor) locus to the broken (recipient) locus, a process termed gene conversion 36.
HR initiates with extensive 5′ to 3′ end-processing at broken ends, which in yeast is regulated by Mre11/Rad50/Xrs2 (MRX, the functional homolog of MRN), Exo1, and at least one other exonuclease 37. The resulting 3′ ssDNA tails are bound by RPA, which is replaced with Rad51 in a reaction mediated by Rad52 and two Rad51 paralogs, Rad55 and Rad57. The resulting Rad51 nucleoprotein filament searches for and invades a homologous sequence, a process facilitated by Rad54. The Srs2 helicase is thought to dissociate Rad51 from ssDNA, allowing normal base-pairing of the invading and complementary donor strands and subsequent strand extension by DNA polymerase. The extended strand can dissociate and anneal with the processed end of the non-invading strand on the opposite side of the DSB in a process called synthesis-dependent strand annealing (SDSA), or both ends may invade producing a double-Holliday junction that is resolved to yield crossover or non-crossover recombinants. Once intermediates are resolved, the remaining ssDNA gaps and nicks are repaired by DNA polymerase and DNA ligase. As with NHEJ, most HR proteins are conserved through evolution, although mammals harbor a more elaborate set. For example, there are five Rad51 paralogs in mammals (Rad51B/C/D and XRCC2/3) but just two in yeast (Rad55/57). During meiotic HR, the Rad51 homolog Dmc1 participates in strand exchange with Rad51, an association conserved from yeast to human. Several mammalian HR proteins do not have homologs in yeast, including BRCA1 and BRCA2. As with NHEJ, the role of BRCA1 in HR is unclear, but it interacts with BRCA2 and functions within the Fanconi's anemia protein ubiquitylation pathway that may regulate protein-protein interactions and/or accessibility of repair factors to damage sites 29. More detailed information about HR protein biochemistry and repair mechanisms is available in several recent reviews 2, 36, 37, 38 and in the article by Li and Heyer in this issue.
Crossovers are associated with a fraction of HR events and can have a stabilizing or destabilizing effect on the genome. In meiosis, crossovers are highly regulated such that at least one crossover occurs between each pair of homologous chromosomes to ensure proper chromosome segregation, yet excess crossovers are suppressed 39. In mitosis, crossovers pose serious risks of large-scale genome alterations: half of the G2 phase crossovers between homologs result in loss of heterozygosity from the point of the crossover to the telomere, and crossovers between repeated regions on non-homologous chromosomes, the same chromosome, or sister chromatids can result in translocations, inversions, deletions, and gene duplications 36. Defects in proteins that suppress mitotic crossovers, such as Sgs1 in yeast and its human homolog BLM, increase genome instability, and BLM defects also predispose to cancer 40, 41.
Yeast mutants lacking key HR proteins (i.e., Mre11, Rad51, Rad52, Rad54) are viable, but DSBs often go unrepaired and this results in cell death in haploids. Diploids usually survive the loss of a broken chromosome, or a full chromosome complement can be retained if a single end invades the homologous chromosome and primes repair synthesis to the end of the chromosome (> 100 Kb distant in some cases), a process called “break-induced replication” that results in large-scale loss of heterozygosity 42, 43. In contrast, loss of key HR proteins in higher eukaryotes, including RAD51, BRCA1, and BRCA2, results in cell and/or embryonic lethality; viable mutants in these cases typically carry hypomorphic alleles or lethality is suppressed by p53 mutations. In other cases, as with mutations in higher eukaryotic HR proteins (e.g., RAD52 and RAD54), the HR defects are milder than those of the corresponding yeast mutant 44, 45, 46. Although it has been argued that these differences may reflect changes in the functions of these proteins through evolution 47, we would argue that the greater requirement for RAD51 and BRCA1/2 in higher eukaryotes reflects the essential role of HR in restarting blocked or collapsed replication forks, which is probably required 100-fold more often in higher eukaryotes than in yeast because higher eukaryotic genomes are 100-fold larger than the yeast genome. The reduced requirement for certain HR proteins in higher eukaryotes may reflect diverged functions and/or functional redundancy with the elaboration of HR protein families and addition of new HR proteins, as proposed for RAD52 and BRCA2 48.
Although NHEJ is responsible for the vast majority of tumorigenic chromosomal translocations 49, and even “correct” re-joining of broken ends by NHEJ often results in mutations at junctions, the NHEJ machinery plays a significant role in maintaining genome stability and suppressing tumorigenesis 50, 51, 52, 53, 54. These results indicate that NHEJ is not indiscriminate, but instead mediates DSB repair with a fair degree of fidelity. The genome-stabilizing and tumor-suppressing functions of the HR machinery are similarly well established 36. Thus, both DSB repair pathways play critical roles in maintaining genome stability and preventing cancer. In the following sections we discuss how cells modulate the relative levels of the two DSB repair pathways, and their respective fidelities, to maintain an appropriate level of genome stability. Several factors are important in this regulation, including the availability of repair templates, cell cycle phase, proliferation rate, and the functions of specific cell types.
DSB repair pathway choice in yeast and higher eukaryotes
Early studies of cell killing by IR gave rise to the notion that HR is the dominant mechanism of DSB repair in yeast. Yeast has very limited capacity for imprecise NHEJ, which is the main option for NHEJ repair of “dirty” ends produced by IR, as these ends require processing before rejoining in a process regulated by the MRX complex. Note that MRX also regulates the more extensive end-processing that resects broken ends to long 3′ tails early in HR; this resection is slowed but not eliminated in mre11 mutants 55, 56, or in mre11 exo1 double mutants, indicating that at least one other nuclease is involved 37. MRX plays a critical role in processing IR-induced DSBs, and in removing Spo11 nuclease, which remains covalently bound to broken ends after it creates meiotic DSBs. Thus, both IR-induced HR and meiotic HR are markedly reduced in mre11 mutants 57, 58. In contrast, mre11 mutants show relatively mild (< 2-fold) defects in HR stimulated by “clean” DSBs produced by HO nuclease 43, 55, 59, indicating that the low level of IR-induced HR in mre11 mutants does not reflect an HR defect per se. By contrast, mre11 mutation decreases (precise) NHEJ of HO-induced DSBs by ∼100-fold 60, 61. When HR is blocked and yeast cells are forced to repair a nuclease DSB by imprecise NHEJ, cell survival is a measure of such repair, which ranges from 0.01% to 0.2% 14, 61, 62. In this assay, HO nuclease is expressed continuously; precise repair recreates the HO recognition site, which gets cleaved again; so cells are forced to repair DSBs by imprecise NHEJ to break the futile cycle. The low efficiency of imprecise NHEJ contrasts with the robust capacity of yeast to repair these DSBs by precise NHEJ, which we estimated at a minimum of 25% based on the increase in HR when NHEJ was blocked by _yku70_Δ mutation 14; the remaining DSBs are repaired by the robust HR pathway.
It is not known why imprecise NHEJ is so inefficient in yeast compared to mammals, but the most likely reason is that mammals have at least three NHEJ proteins that are absent in yeast: DNA-PKcs, BRCA1, and Artemis. We propose that DNA-PKcs is the key missing protein because it facilitates alignment of non-complementary ends and regulates end-processing during NHEJ 63, 64; BRCA1 has broader roles in DNA repair as well as roles in transcription and other cellular functions 65, 66, and Artemis has specialized roles in processing a small subset of broken ends 25.
Both precise and imprecise NHEJ are robust in higher eukaryotes. It can be difficult to distinguish precise NHEJ in a chromosomal context from the failure to induce a DSB, and precise NHEJ of transfected linear plasmids has often been used as a surrogate. The Waldman lab devised a clever assay with an integrated substrate carrying two I-_Sce_I nuclease recognition sites to show that the frequency of precise NHEJ is ∼50% in mouse cells 15. Naturally, imprecise NHEJ predominates for IR-induced DSBs, although this is strongly affected by cell cycle phase (see below). The imprecision of NHEJ during V(D)J recombination, initiated by RAG1/2 nuclease-induced DSBs, plays a major role in generating antibody diversity in mammals 67. It is likely that robust imprecise NHEJ repair systems were a key driving force behind the evolution of adaptive immune systems.
The greater use of imprecise NHEJ in higher eukaryotes may also be related to their larger genome size. Although random small-scale deletions and insertions may have a lesser chance of affecting coding sequences in a mammalian genome (comprising only ∼3% of the genome) than in a yeast genome (> 70% coding), it is now recognized that as much as 50% of mammalian genomes are transcribed into functional RNAs, including many microRNAs that regulate gene expression. The larger genomes of higher eukaryotes present a greater challenge of locating a homologous template for HR repair. However, it is remarkable that HR between ectopic loci, and between allelic loci on homologous chromosomes, is very efficient (albeit quite slow) even in the cells with the largest genomes. Nuclear architecture appears to confine chromosomes to well-defined territories, both in yeast and in mammalian cells 68, 69. Although homologs are usually not in close proximity, chromosome territories could limit the “search space” and increase the efficiency of HR.
Differential DSB repair pathway choices in different cell types of a single species
A key factor that regulates HR efficiency is template availability; thus, it is not surprising that HR is more efficient in diploid than in haploid yeast. However, the relationship between ploidy and HR efficiency in yeast is not simple. It has been known for almost 50 years that tetraploids are more radiosensitive than diploids, and diploids that express both MAT alleles (MAT a and _MAT_α – termed a/α) are more radioresistant than diploids that express only one copy of MAT (a/a or α/α diploids) 70. These and other results indicate that HR efficiency is independently regulated by ploidy and the MAT loci 71, 72, 73. Enhanced HR with greater availability of homologous templates indicates that template accessibility is rate limiting for HR. Of course, template availability increases in all cells when DNA is replicated in S phase; this factor is discussed in the section on cell cycle regulation below.
The mechanisms by which MAT heterozygosity enhances HR are still under investigation. One way to enhance HR is to suppress or genetically inactivate NHEJ, as seen in yeast and mammalian cells 14, 74, 75, 76, and this strategy is used by wild-type yeast: the Nej1 protein interacts with Lif1 and enhances the ligase activity of the Dnl4/Lif1 complex, but Nej1 is expressed only in haploids because NEJ1 is repressed by MAT a1/α2, the transcriptional repressor expressed in (MAT heterozygous) diploids. _nej1_Δ mutants show an NHEJ defect as strong as NHEJ mutants yku70/80, dnl4, and lif1 77, 78, 79, and the suppression of Nej1 expression in diploids decreases NHEJ by ∼10-fold. The expression of other NHEJ proteins does not appear to be regulated by MAT. Thus, an important mechanism by which haploids upregulate NHEJ and downregulate HR (and vice versa in diploids) is through _MAT_-dependent regulation of Nej1, and the variable contributions of NHEJ and HR in haploid and diploid yeast reflect independent changes in both DSB repair pathways.
A recent study showed that mutations in a number of genes regulated by the MAT a1/α2 repressor (and other genes not regulated by MAT) suppressed the hypersensitivity of specific rad51, rad52, and rad55 mutants to DNA damage induced by camptothecin and phelomycin. These suppressor mutations occurred in genes that encode NHEJ proteins like Nej1, the meiosis repressor Rme1 and its co-regulator Sin3, chromatin-associated proteins Pst2 and Rfs1, and an unknown protein Ygl193c 80. Interestingly, these suppressor mutations acted differently for each of the HR protein defects, suggesting an extremely complex regulatory network. Note that some or perhaps most of these suppressor mutations do not act by directly regulating HR, particularly mutations in NHEJ genes. For example, the enhanced survival of _rad55_Δ _nej1_Δ relative to _rad55_Δ after treatment with camptothecin may reflect a reduction in lethal chromosome fusions mediated by NHEJ repair of the many DSBs that form when replication forks encounter DNA lesions 80.
HR plays a relatively minor role in DSB repair in many but not all types of higher eukaryotic cells. For example, chicken B lymphocytes generate antibody diversity via DSB repair by gene conversion rather than by imprecise NHEJ during V(D)J recombination as is common in mammals, so it is not surprising that HR is more robust in chicken B cells than in most types of mammalian cells. The high HR capacity of chicken B cells allows efficient HR-mediated gene targeting and gene replacement, and this has propelled chicken B cells, exemplified by the DT-40 system, to the forefront of vertebrate somatic cell genetics 81.
Mouse embryonic stem (ES) cells also display enhanced HR capacity that facilitates gene targeting and construction of novel mutant mice. A plausible explanation for the hyper-recombination phenotype of mouse ES cells is based on the observation that these rapidly dividing cells, while genotypically p53+, are functionally p53− (or p53-suppressed) with respect to DNA damage responses 82. Importantly, p53 is a well-known suppressor of HR 83, 84, 85, 86, 87, 88; thus, the functional inactivation of p53 in ES cells can explain their increased HR capacity. The p53 DNA damage response may be suppressed in ES cells to prevent cell cycle arrest during the early stages of development, which require rapid cell division. Note that ES cells retain a p53-independent apoptotic response pathway to rid the organism of cells with damaged genomes. It is equally plausible (and not mutually exclusive) that p53 is suppressed to enhance HR so that stalled or collapsed replication forks are restarted in a timely manner during the rapid cell divisions in early embryogenesis, and perhaps also to upregulate accurate DSB repair to help maintain genome integrity during this critical developmental stage.
Recent evidence indicates that NHEJ and HR are modulated during different stages of nervous system development in mice. This was inferred from genetic analysis showing that HR defects increased apoptosis in proliferating neural precursor cells and NHEJ defects increased apoptosis later in development, in differentiating cells 89. The lack of effect of NHEJ defects during early development of the nervous system suggests not only that HR activity is enhanced, but also that HR is the predominant mode of DSB repair at this stage of development 89. This suggests that there may be active suppression of NHEJ during early neural development. The stronger role for HR in proliferating nervous tissue early in development is reminiscent of the situation in ES cells noted above.
Cell-cycle regulation of DSB repair pathways
It has been known for some time that the balance between NHEJ and HR shifts during the cell cycle. Recent studies have begun to clarify the molecular mechanisms that regulate these cyclical shifts in the two repair pathways (summarized in Figure 2). Because template accessibility influences HR efficiency, it is not surprising that cells upregulate HR during S and G2 phases of the cell cycle when sister chromatids are available. In fact, sister chromatids are the preferred template for HR repair in yeast and mammalian cells 44, 90. This preference probably reflects a proximity effect mediated by the close association of sister chromatids from the time they form in S phase until they segregate in anaphase. Sister chromatid cohesion is mediated by cohesins, and recent evidence indicates that cohesins migrate to DSB repair sites independently of the normal replication cycle 91. Sister chromatid cohesion is a property of all eukaryotic cells. Yet, NHEJ remains active throughout S and G2 phases of the cell cycle 92. This indicates that NHEJ competes for DSBs even when homologous templates are in the immediate vicinity.
Figure 2
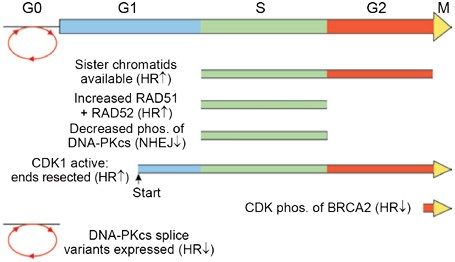
Cell cycle regulation of DSB repair pathway choice. The cell cycle is shown at the top and each line below indicates a specific mechanism that regulates NHEJ or HR in the indicated cell cycle phases. G0 indicates quiescent (non-cycling) cells.
Increasing evidence indicates that the shift from NHEJ toward HR as cells progress from G1 to S/G2 is actively regulated in lower and higher eukaryotes. Early studies showed that RAD51 and RAD52 expression increases during S phase 93. Cyclin-dependent kinases (CDKs) are key regulators of cell cycle progression. In Saccharomyces cerevisiae, the Cdc28 (CDK1) kinase is activated at a point in G1 phase called “Start” which commits to progress to S phase 94. Ira et al. 95 analyzed resection of HO-induced DSBs at MAT and found that G1-arrested cells failed to initiate efficient end resection, which prevented loading of RPA and Rad51, and blocked Mec1 activation. Resection depends on CDK1 activity because suppression of CDK1 in G2 cells by overexpression of the CDK1 inhibitor Sic1, or by use of a mutant form of CDK1 blocked by the ATP analog 1-NMMPP1, also prevented end-resection and checkpoint activation. Interestingly, blocking CDK1 led to the persistence of Mre11 at the DSB site, suggesting that CDK1 activity is not required for MRX recruitment to broken ends, but is required for MRX regulation of end resection 95. The Rad51 nucleoprotein filament is required for the homology search and strand invasion steps of HR. Thus, HR is blocked when ssDNA resection is prevented by inhibition of CDK1, and in early G1 when CDK1 is inactive.
Aylon et al. 96 reached similar conclusions using cells arrested in G1 with α-factor, a temperature-sensitive cdc4 mutant that cannot initiate replication, and in cells treated with hydroxyurea, which blocks replication. In all cases HR was defective in the arrested cells. However, HR was observed in a significant fraction of a cdc7 mutant population. The cdc7 mutation prevents replication initiation, but not progression through later cell cycle phases in cells shifted to the restrictive temperature after replication has initiated. The fact that HR is blocked even in G1-arrested diploid cells argues that HR is tightly regulated by CDK-dependent cell cycle controls, and that the presence of a homologous template is not sufficient for HR competence. Given the suppression of HR in early G1, it is not surprising that G1-arrested budding yeast cells are capable of repairing DSBs by NHEJ 96, and similar results were obtained in fission yeast 97. Caspari et al. 98 identified additional CDK-mediated HR regulatory mechanisms in fission yeast, showing that the Cdc2-cyclin B CDK is important in early HR when Rhp51 (Rad51 homolog) is assembled onto ssDNA, and at a late HR stage in which topoisomerase III helps resolve HR intermediates.
CDKs also regulate HR in mammalian cells. The West laboratory showed that CDK-mediated phosphorylation of serine 3291 of BRCA2 blocks the interaction of RAD51 with this C-terminal region of BRCA2 99. More recently, this group has shown that this phospho-regulated binding site for RAD51 recognizes RAD51 in its multimeric forms, including DNA-bound RAD51 nucleoprotein filaments and DNA-free multimers 100. This particular phosphorylation is maximal in M phase and therefore represents one of the mechanisms by which HR is downregulated in M and early G1 phase. Interestingly, this cell cycle control of HR was bypassed when cells were irradiated, as this led to a rapid decrease in serine 3291 phosphorylation and increased association of BRCA2 with RAD51 to promote DSB repair by HR. Although not directly addressed experimentally, it seems likely that the decreased serine 3291 phosphorylation after IR is cell cycle independent, i.e., the DNA damage response network is able to bypass the normal cell cycle control of the BRCA2-RAD51 interaction.
A key requirement for mammalian NHEJ is the phosphorylation of clusters of serine and threonine residues in DNA-PKcs, targeted by DNA-PKcs itself and ATM. DNA-PKcs is _trans_-autophosphorylated at a cluster of six residues that includes T2609 (also called “ABCDE”) 101, 102, at a separate cluster that includes S2056 (also called “PQR”) 18, 103, and at T3950 104. T2609 is also phosphorylated by ATM – see below. DNA-PKcs kinase activity and phosphorylation of T2609 and T3950 are critical for NHEJ and cellular radioresistance 102, 105, 106. Phosphorylation of the S2056 and T2609 clusters is reduced in irradiated S phase cells 103, and this may be part of a regulatory system that downregulates NHEJ in S phase. This model is consistent with biochemical assays showing decreased DNA-PK activity in S phase HeLa cells 107.
Proteins at the HR/NHEJ interface
The idea that the two DSB repair pathways compete for DSBs was suggested by early experiments showing that plasmid DNA transfected into mammalian cells could be repaired by either pathway 108, and biochemical studies showing both NHEJ and HR proteins bind to broken ends [reviewed in refs. 109, 110]. The increase in HR seen in cells with NHEJ defects is consistent with passive shunting of DSBs from NHEJ to HR 14, 74, 75, 76. However, it was recognized early that yeast Mre11 influenced both pathways 111, raising the possibility that the competition between the two pathways might be actively controlled, and several other proteins are now implicated in both pathways, including BRCA1, histone H2AX, PARP-1, RAD18, DNA-PKcs, and ATM. Although NHEJ factors are recruited to DSBs more rapidly than HR factors, and NHEJ and HR factors are independently recruited to DSBs, there is a significant period of time when both sets of factors are present at damage sites 112, consistent with the notion that pathway choice may be regulated by one or more proteins that act in both pathways.
As mentioned above, BRCA1 involvement in HR remains unclear but may reflect functional interactions with BRCA2, a critical mediator of the RAD51 strand transferase 66, 113, 114, 115, and its role in the Fanconi's anemia ubiquitylation pathway 29. It is not clear how BRCA1 promotes NHEJ, although a study showing that BRCA1 negatively regulates end-processing by MRE11 endo- and exonucleases 116 suggests a plausible mechanism by which BRCA1 can influence both pathways. With respect to NHEJ, BRCA1 suppression of MRN-mediated end-processing may enhance NHEJ accuracy [reviewed in refs. 28, 115]. A recent report demonstrates that Chk2 phosphorylation of BRCA1 influences the fidelity of NHEJ 117. Together, the results indicate that BRCA1 promotes genome stability by promoting error-free HR and by maximizing the fidelity of NHEJ. Because BRCA1 functions in both DSB repair pathways, it is possible that BRCA1 regulates pathway choice, but as yet there is no direct evidence to support this idea.
Histone H2AX is a subunit of the nucleosome that is rapidly phosphorylated over megabase domains at DSBs in mammalian chromatin, principally by ATM and ATR. The yeast H2AX homolog, H2A, is phosphorylated over kilobase domains at DSBs by the ATM and ATR homologs Tel1 and Mec1. H2AX phosphorylation plays a key role in DNA damage checkpoint activation, and its dephosphorylation is important for attenuating the checkpoint signal to allow the cell cycle to resume (118, see also Huen and Chen in this volume). Mutation of the phosphorylated serine 129 residue in yeast H2A causes sensitivity to DNA-damaging agents and confers an NHEJ defect 119. A recent study showed that deletions or amino acid substitutions in both C- and N-terminal H2A tails confer an NHEJ defect 120. However, the nature of NHEJ defect is unclear because it was revealed in an assay involving rejoining of a transfected plasmid, which may not be chromatinized prior to repair. Mutation of the analogous residue in mouse H2AX (serine 139) confers a defect in conservative DSB repair by gene conversion 121. Although direct measures of HR in yeast H2A mutants have not been reported, H2A phosphorylation is important for recruitment or retention of checkpoint and other repair factors to DSBs. For example, recruitment of the INO80 chromatin remodeling complex depends on H2A phosphorylation, and deletion of the INO80 subunit Arp8 results in ∼4-fold reductions in HO-nuclease-induced MAT switching in haploids and allelic HR in diploids (Y-C Lo, T Tsukuda, R Sterk, S Krishna, MA Osley, JA Nickoloff, unpublished results). A recent study identified an interaction between the mouse Polycomb Group YY1 transcription factor and the INO80 chromatin remodeling complex, and showed that siRNA knockdown of either YY1 or the Ino80 subunit caused a specific defect in HR repair of DSBs [122](/articles/cr2007111#ref-CR122 "Wu S, Shi Y, Mulligan P, et al. A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nat Struct Mol Biol 2007 Nov 18; doi: 10.1038/nsmb1332
."). It will be interesting to determine whether mammalian chromatin remodeling complexes like YY1-INO80 also regulate NHEJ. The roles of chromatin modification and remodeling in DNA repair were recently reviewed ([123](/articles/cr2007111#ref-CR123 "Osley MA, Tsukuda T, Nickoloff JA . ATP-dependent chromatin remodeling factors and DNA damage repair. Mutat Res 2007; 618:65–80."), [124](/articles/cr2007111#ref-CR124 "Downs JA, Nussenzweig MC, Nussenzweig A . Chromatin dynamics and the preservation of genetic information. Nature 2007; 447:951–958."), see also the article by Huen and Chen in this issue).PARP-1 functions with Ligase III in an alternative, lower fidelity NHEJ pathway, and PARP-1 competes with Ku for DSB ends 30. Interestingly, PARP-1 defective DT-40 cells reportedly show reduced HR levels, inferred from camptothecin hypersensitivity, and normal resistance to camptothecin was restored in the PARP-1 defect cells by mutations that inactivate NHEJ 125. Defects in RAD18 showed the same hypersensitivity and genetic interactions with NHEJ, and the double PARP-1/RAD18-defective mutant showed synergistic hypersensitivity to camptothecin, suggesting that RAD18 and PARP-1 independently promote HR and antagonize NHEJ 125; however, a conflicting result was obtained in mouse ES cells 126. Here, the PARP-1 mutant showed enhanced HR measured directly as enhanced HR-mediated gene targeting. While the reason for this discrepancy is not clear, these studies implicate PARP-1 (and RAD18) in regulating DSB repair pathway choice.
As mentioned above, increased HR in cells with NHEJ defects can be explained by a passive competition model. However, several lines of evidence suggest that DNA-PKcs is an active regulator of DSB repair pathway choice. The first evidence for this was the puzzling finding that inactivation of NHEJ by elimination of DNA-PKcs increased HR 74, but chemical inhibition of DNA-PKcs had the opposite effect 127. Phosphorylation of DNA-PKcs is important for NHEJ 105, and biochemical experiments indicate that phosphorylation of the T2609 cluster causes DNA-PKcs to dissociate from broken ends 106, 128, 129, 130, 131, 132, 133. Together, these results suggested a model in which chemically inhibited DNA-PKcs fails to dissociate from ends and thereby blocks access to other NHEJ repair factors and to HR repair factors. The inhibition of both DSB repair pathways by the specific inhibition of DNA-PKcs has important implications for tumor radiosensitization 127. The importance of phosphorylation of DNA-PKcs for HR was underscored by a study showing that complementation of DNA-PKcs null CHO V3 cells with DNA-PKcs lacking the T2609 cluster sites gave the same phenotype as chemical inhibition of wild-type DNA-PKcs, i.e., HR was suppressed. Thus, blocking phosphorylation of the T2609 cluster converts DNA-PKcs into a dominant-negative regulator of HR 18.
The idea that DNA-PKcs regulates DSB repair pathway choice gained additional support when the Meek laboratory identified DNA-PKcs splice variants that lack the kinase domain 134. Given the importance of DNA-PKcs kinase activity for NHEJ 105, it was not surprising that these kinase-inactive variants do not complement the radiosensitivity of DNA-PKcs null cells. However, the variants were particularly interesting because they had dominant negative effects on HR repair of DSBs, and therefore mimicked chemically inhibited full-length DNA-PKcs and the T2609 cluster mutant. The variants are expressed along with full-length DNA-PKcs only in quiescent cells, but they do not have a dominant negative effect on NHEJ. These results suggest that co-expression of full-length DNA-PKcs and kinase-inactive variants limits HR in quiescent cells, which lack preferred sister chromatid HR repair templates, while simultaneously maintaining a high capacity for DSB repair by NHEJ 134. These results strongly support the notion that DNA-PKcs is a key regulator of DSB repair pathway choice in higher eukaryotes.
Co-regulation of HR by DNA-PKcs and ATM
HR is stimulated when DNA-PKcs is absent, but repressed when DNA-PKcs is chemically inhibited, harbors mutations in the T2609 autophosphorylation cluster, or is expressed as a kinase-inactive splice variant. We therefore reasoned that expression of a kinase-inactive mutant of DNA-PKcs (harboring a single lysine to arginine change near the kinase active site, K3752R) would similarly act as a dominant negative regulator of HR. Surprisingly, when CHO V3 cells were complemented with the K3752R mutant protein, HR was stimulated, with levels ∼3-fold above the already elevated HR level seen in DNA-PKcs null cells (manuscript submitted). We used immunofluorescence microscopy to demonstrate that DNA-PKcs recruitment to foci after IR occurs independently of its kinase activity, suggesting that the kinase-inactive K3752R mutant somehow upregulates HR despite its association with broken ends and its inability to be autophosphorylated.
Several lines of evidence help explain the hyper-recombination phenotype of the K3752R mutant. First, ATM levels are reduced in cells lacking DNA-PKcs 135, but we found that ATM levels are restored in DNA-PKcs null cells upon expression of the DNA-PKcs K3752R mutant protein. Thus, DNA-PKcs regulates ATM levels independently of DNA-PKcs kinase activity. Second, two studies have shown that the DNA-PKcs T2609 cluster is also phosphorylated by ATM 102, 136. DNA-PKcs has another cluster of phosphorylation sites that includes S2056, and phosphorylation of the T2609 and S2056 clusters regulates accessibility of repair factors to DSBs 18, 106, 128, 129, 130, 131, 132, 133, but, unlike the T2609 cluster, the S2056 cluster is subject only to autophosphorylation 136. Thus, in cells expressing DNA-PKcs K3752R, the S2056 cluster is not phosphorylated. Mutations in the S2056 cluster that block phosphorylation increase DSB-induced HR above the wild-type level, but not above the level seen in DNA-PKcs null cells 18. Thus, the higher HR levels with DNA-PKcs K3752R cannot simply be due to failure to phosphorylate the S2056 cluster, but probably reflect the combined positive effects of preventing S2056 cluster phosphorylation, and the absence of competition by NHEJ. Presumably, the kinase-inactive DNA-PKcs K3752R mutant still needs to be released from broken ends by T2609 cluster phosphorylation, which is mediated by ATM 102, 136. In this model, ATM-mediated phosphorylation of the T2609 cluster is one mechanism by which ATM promotes HR in cells expressing the DNA-PKcs K3752R mutant. Note that HR is strongly reduced by chemical inhibitors of DNA-PKcs and by kinase-inactive DNA-PKcs splice variants, but HR is strongly enhanced by kinase-inactive DNA-PKcs harboring the single amino acid K3752R mutation. One possibility is that DNA-PKcs adopts a conformation that blocks ATM phosphorylation of the T2609 cluster when inhibitors are bound to the DNA-PKcs active site, and when the protein lacks the entire kinase domain. The K3752R mutation, on the other hand, would be expected to have little or no effect on the gross structure of DNA-PKcs, and the mutant protein should therefore be recognized and phosphorylated by ATM in the same way as wild-type DNA-PKcs.
A second independent mechanism of HR promotion by ATM is through phosphorylation of other targets. ATM phosphorylates many targets, at least 12 of which have direct or indirect roles in HR (Figure 3A). Interestingly, 6 of these targets are also phosphorylated by DNA-PKcs. The role of ATM in HR has been puzzling. Spontaneous HR is enhanced in ATM-defective cells 137, but this may reflect a replication- or checkpoint-specific effect. For DSB-induced HR, ATM appears to have a positive role 138, 139, perhaps through phosphorylation of H2AX 121. However, direct measurements of DSB-induced HR showed no differences between wild-type and ATM-defective cells 140, 141, perhaps because DNA-PKcs fulfilled this role. Note that DNA-PKcs and ATM do not show simple redundancy with regard to H2AX phosphorylation. DNA-PKcs can phosphorylate H2AX in the absence of ATM, but not in its presence, or even in the presence of kinase-inactive ATM, suggesting that ATM physically blocks DNA-PKcs access to H2AX [reviewed in ref. 24]. Defects in mouse ATM and DMC1 confer similar meiotic HR defects 142. Caffeine inhibits ATM (and other PI3 kinases) and markedly reduces DSB-induced HR 143, and we also found that the ATM-specific inhibitor KU55933 144 blocks formation of IR-induced RAD51 foci (manuscript submitted). Together, these results support the idea that ATM promotes HR repair of DSBs. A positive role for ATM in HR can also explain the puzzling finding that DNA-PKcs null cells are hypersensitive to replication-blocking agents, which was taken as evidence of a limited role for NHEJ in restarting collapsed replication forks 145. However, because DSEs at collapsed forks have no second end with which to rejoin (Figure 1A), we propose an alternative explanation, that in the absence of DNA-PKcs the reduced levels of ATM 135 inhibit HR-mediated replication fork restart.
Figure 3
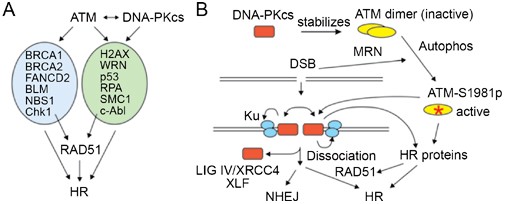
Model for co-regulation of NHEJ and HR by DNA-PKcs and ATM. (A) Lists of proteins involved in HR that are phosphorylated by ATM, and the subset that are phosphorylated by DNA-PKcs. ATM also phosphorylates DNA-PKcs. (B) DNA-PKcs stabilizes ATM, shown at the top as an inactive dimer. DSBs lead to MRN recruitment, ATM autophosphorylation on serine 1981, and dissociation into active monomers. The Ku heterodimer also arrives early at DSBs and recruits DNA-PKcs, which becomes activated upon DNA end-binding and phosphorylates itself, Ku, and other proteins. DNA-PKcs is phosphorylated by itself and ATM, causing DNA-PKcs to dissociate from ends and allowing access to NHEJ and HR proteins. In the absence of DNA-PKcs, ATM levels drop and HR is mildly enhanced because NHEJ no longer competes for DSBs. When DNA-PKcs is chemically inhibited, it remains bound to ends, blocking access to both HR and NHEJ factors. When DNA-PKcs harbors a point mutation in the kinase domain, it no longer supports NHEJ but does stabilize ATM, and ATM is able to phosphorylate its T2609 cluster (and all HR factors in panel A), greatly increasing HR.
In summary, HR is elevated in cells expressing DNA-PKcs K3752R and DNA-PKcs null cells because NHEJ competition is eliminated, and we propose that HR levels are highest in the DNA-PKcs K3752R mutant because the mutant protein restores ATM to its normal level and ATM is thus able to exert its full positive effect on HR, including phosphorylation of a large number of HR proteins, and the DNA-PKcs T2609 cluster to facilitate access of HR proteins to the broken ends (Figure 3B). ATM is also a positive regulator of NHEJ after IR 146, which reflects at least in part the phosphorylation of Artemis 25, and ATM has an important role in stabilizing broken ends during NHEJ-mediated V(D)J recombination 147, but these functions are unlikely to impact HR induced by nuclease DSBs.
Potential benefits of DSB repair pathway analysis
As we gain a better understanding of DSB repair mechanisms and the regulation of pathway choice, it is likely that basic mechanistic insights will translate into clinical benefits. The complex network of DSB repair proteins and the regulatory proteins including the PI3 kinases DNA-PKcs, ATM and ATR represent a rich set of potential targets to exploit in the development of more effective chemo- and radiotherapeutic strategies in cancer therapy. These targets may also be useful as biomarkers of genome instability to improve our ability to detect cancer in its earliest stages when treatments are most effective. Human gene therapy is currently hindered by risks associated with random integration of transgenes, with significant potential for insertional mutagenesis of tumor suppressors or inappropriate activation of oncogenes. Targeted gene therapy, including accurate gene replacement and transgene insertion into low-risk regions of the genome, depends on suppressing NHEJ-mediated random integration and enhancing HR-mediated integration into desired loci. The proteins that regulate the choice between NHEJ and HR are excellent targets to manipulate to enhance gene targeting and unleash the full potential of targeted gene therapy with minimum risk of untoward side effects.
References
- Shen Z, Nickoloff JA . Mammalian homologous recombination repair and cancer intervention. In: Wei Q, Li L, Chen DJ, eds. DNA Repair, Genetic Instability, and Cancer. Singapore: World Scientific Publishing Co., 2007:119–156.
Chapter Google Scholar - Paques F, Haber JE . Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 1999; 63:349–404.
CAS PubMed PubMed Central Google Scholar - Franco S, Alt FW, Manis JP . Pathways that suppress programmed DNA breaks from progressing to chromosomal breaks and translocations. DNA Repair (Amst) 2006; 5:1030–1041.
Article CAS Google Scholar - Chaudhuri J, Basu U, Zarrin A, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol 2007; 94:157–214.
Article CAS PubMed Google Scholar - Keeney S, Neale MJ . Initiation of meiotic recombination by formation of DNA double-strand breaks: mechanism and regulation. Biochem Soc Trans 2006; 34:523–525.
Article CAS PubMed Google Scholar - Murnane JP . Telomeres and chromosome instability. DNA Repair (Amst) 2006; 5:1082–1092.
Article CAS Google Scholar - Acilan C, Potter DM, Saunders WS . DNA repair pathways involved in anaphase bridge formation. Genes Chromosomes Cancer 2007; 46:522–531.
Article CAS PubMed Google Scholar - Ward J . The nature of lesions formed by ionizing radiation. In: Nickoloff JA, Hoekstra MF, eds. DNA Damage and Repair: DNA Repair in Higher Eukaryotes. Totowa, NJ: Humana Press, 1998:65–84.
Google Scholar - Limoli CL, Giedzinski E, Bonner WM, Cleaver JE . UV-induced replication arrest in the xeroderma pigmentosum variant leads to DNA double-strand breaks, γ-H2AX formation, and Mre11 relocalization. Proc Natl Acad Sci USA 2002; 99:233–238.
Article CAS PubMed Google Scholar - Bosco EE, Mayhew CN, Hennigan RF, et al. RB signaling prevents replication-dependent DNA double-strand breaks following genotoxic insult. Nucleic Acids Res 2004; 32:25–34.
Article CAS PubMed PubMed Central Google Scholar - Degrassi F, Fiore M, Palitti F . Chromosomal aberrations and genomic instability induced by topoisomerase-targeted antitumour drugs. Curr Med Chem Anticancer Agents 2004; 4:317–325.
Article CAS PubMed Google Scholar - Su TT . Cellular responses to DNA damage: one signal, multiple choices. Annu Rev Genet 2006; 40:187–208.
Article CAS PubMed Google Scholar - Rothstein R, Michel B, Gangloff S . Replication fork pausing and recombination or “gimme a break”. Genes Dev 2000; 14:1–10.
CAS PubMed Google Scholar - Clikeman JA, Khalsa GJ, Barton SL, Nickoloff JA . Homologous recombinational repair of double-strand breaks in yeast is enhanced by MAT heterozygosity through yKu-dependent and -independent mechanisms. Genetics 2001; 157:579–589.
CAS PubMed PubMed Central Google Scholar - Lin Y, Lukacsovich T, Waldman AS . Multiple pathways for repair of double-strand breaks in mammalian chromosomes. Mol Cell Biol 1999; 19:8353–8360.
Article CAS PubMed PubMed Central Google Scholar - Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ . ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 2001; 276:42462–42467.
Article CAS PubMed Google Scholar - Collis SJ, DeWeese TL, Jeggo PA, Parker AR . The life and death of DNA-PK. Oncogene 2005; 24:949–961.
Article CAS PubMed Google Scholar - Cui X, Yu Y, Gupta S, et al. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol Cell Biol 2005; 25:10842–10852.
Article CAS PubMed PubMed Central Google Scholar - Shao RG, Cao CX, Zhang H, et al. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J 1999; 18:1397–1406.
Article CAS PubMed PubMed Central Google Scholar - Burma S, Chen DJ . Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst) 2004; 3:909–918.
Article CAS Google Scholar - Chan DW, Ye RQ, Veillette CJ, Lees-Miller SP . DNA-dependent protein kinase phosphorylation sites in Ku 70/80 heterodimer. Biochemistry 1999; 38:1819–1828.
Article CAS PubMed Google Scholar - Karmakar P, Piotrowski J, Brosh RM, et al. Werner protein is a target of DNA-dependent protein kinase in vivo and in vitro, and its catalytic activities are regulated by phosphorylation. J Biol Chem 2002; 277:18291–18302.
Article CAS PubMed Google Scholar - Yannone SM, Roy S, Chan DW, et al. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J Biol Chem 2001; 276:38242–38248.
CAS PubMed Google Scholar - Stucki M, Jackson SP . γH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 2006; 5:534–543.
Article CAS Google Scholar - Riballo E, Kuhne M, Rief N, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to γ-H2AX foci. Mol Cell 2004; 16:715–724.
Article CAS PubMed Google Scholar - Jeggo PA, O'Neill P . The Greek goddess, Artemis, reveals the secrets of her cleavage. DNA Repair (Amst) 2002; 1:771–777.
Article CAS Google Scholar - Nick McElhinny SA, Havener JM, Garcia-Diaz M, et al. A gradient of template dependence defines distinct biological roles for family X polymerases in nonhomologous end joining. Mol Cell 2005; 19:357–366.
Article CAS PubMed Google Scholar - Durant ST, Nickoloff JA . Good timing in the cell cycle for precise DNA repair by BRCA1. Cell Cycle 2005; 4:1216–1222.
Article CAS PubMed Google Scholar - Huang TT, D'Andrea AD . Regulation of DNA repair by ubiquitylation. Nat Rev Mol Cell Biol 2006; 7:323–334.
Article CAS PubMed Google Scholar - Wang M, Wu W, Wu W, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res 2006; 34:6170–6182.
Article CAS PubMed PubMed Central Google Scholar - Daley JM, Palmbos PL, Wu D, Wilson TE . Nonhomologous end joining in yeast. Annu Rev Genet 2005; 39:431–451.
Article CAS PubMed Google Scholar - Lees-Miller SP, Meek K . Repair of DNA double strand breaks by non-homologous end joining. Biochimie 2003; 85:1161–1173.
Article CAS PubMed Google Scholar - Burma S, Chen BP, Chen DJ . Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst) 2006; 5:1042–1048.
Article CAS Google Scholar - Rouse J, Jackson SP . Interfaces between the detection, signaling, and repair of DNA damage. Science 2002; 297:547–551.
Article CAS PubMed Google Scholar - Strathern JN, Shafer BK, McGill CB . DNA synthesis errors associated with double-strand break repair. Genetics 1995; 140:965–972.
CAS PubMed PubMed Central Google Scholar - Nickoloff JA . Recombination: mechanisms and roles in tumorigenesis. In: Bertino JR, ed. Encyclopedia of Cancer. 2nd Edition. San Diego, USA: Elsevier Science, 2002:49–59.
Chapter Google Scholar - Krogh BO, Symington LS . Recombination proteins in yeast. Annu Rev Genet 2004; 38:233–271.
Article CAS PubMed Google Scholar - West SC . Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol 2003; 4:435–445.
Article CAS PubMed Google Scholar - Champion MD, Hawley RS . Playing for half the deck: the molecular biology of meiosis. Nat Cell Biol 2002; 4(Suppl):s50–s56.
Article PubMed Google Scholar - Myung K, Datta A, Chen C, Kolodner RD . SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat Genet 2001; 27:113–116.
Article CAS PubMed Google Scholar - Cheok CF, Bachrati CZ, Chan KL, et al. Roles of the Bloom's syndrome helicase in the maintenance of genome stability. Biochem Soc Trans 2005; 33:1456–1459.
Article CAS PubMed Google Scholar - Signon L, Malkova A, Naylor ML, Klein H, Haber JE . Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol Cell Biol 2001; 21:2048–2056.
Article CAS PubMed PubMed Central Google Scholar - Krishna S, Wagener BM, Liu HP, et al. Mre11 and Ku regulation of double-strand break repair by gene conversion and break-induced replication. DNA Repair (Amst) 2007; 6:797–808.
Article CAS Google Scholar - Dronkert MLG, Beverloo HB, Johnson RD, et al. Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange. Mol Cell Biol 2000; 20:3147–3156.
Article CAS PubMed PubMed Central Google Scholar - Essers J, Hendriks RW, Swagemakers SM, et al. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell 1997; 89:195–204.
Article CAS PubMed Google Scholar - Bezzubova O, Silbergleit A, Yamaguchi-Iwai Y, Takeda S, Buerstedde JM . Reduced X-ray resistance and homologous recombination frequencies in a RAD54−/− mutant of the chicken DT40 cell line. Cell 1997; 89:185–193.
Article CAS PubMed Google Scholar - Sonoda E, Hochegger H, Saberi A, Taniguchi Y, Takeda S . Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair (Amst) 2006; 5:1021–1029.
Article CAS Google Scholar - Yang H, Jeffrey PD, Miller J, et al. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 2002; 297:1837–1848.
Article CAS PubMed Google Scholar - Zhang Y, Rowley JD . Chromatin structural elements and chromosomal translocations in leukemia. DNA Repair (Amst) 2006; 5:1282–1297.
Article CAS Google Scholar - Karanjawala ZE, Grawunder U, Hsieh CL, Lieber MR . The nonhomologous DNA end joining pathway is important for chromosome stability in primary fibroblasts. Curr Biol 1999; 9:1501–1504.
Article CAS PubMed Google Scholar - Ferguson DO, Sekiguchi JM, Chang S, et al. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc Natl Acad Sci USA 2000; 97:6630–6633.
Article CAS PubMed PubMed Central Google Scholar - Zha S, Alt FW, Cheng HL, Brush JW, Li G . Defective DNA repair and increased genomic instability in Cernunnos-XLF-deficient murine ES cells. Proc Natl Acad Sci USA 2007; 104:4518–4523.
Article CAS PubMed PubMed Central Google Scholar - Sharpless NE, Ferguson DO, O'Hagan RC, et al. Impaired nonhomologous end-joining provokes soft tissue sarcomas harboring chromosomal translocations, amplifications, and deletions. Mol Cell 2001; 8:1187–1196.
Article CAS PubMed Google Scholar - Tong WM, Cortes U, Hande MP, et al. Synergistic role of Ku80 and poly(ADP-ribose) polymerase in suppressing chromosomal aberrations and liver cancer formation. Cancer Res 2002; 62:6990–6996.
CAS PubMed Google Scholar - Sugawara N, Haber JE . Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol Cell Biol 1992; 12:563–575.
Article CAS PubMed PubMed Central Google Scholar - Lee SE, Moore JK, Holmes A, et al. Saccharomyces Ku70, Mre11/Rad50, and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 1998; 94:399–409.
Article CAS PubMed Google Scholar - Bressan DA, Baxter BK, Petrini JHJ . The Mre11-Rad50-Xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae. Mol Cell Biol 1999; 19:7681–7687.
Article CAS PubMed PubMed Central Google Scholar - Moreau S, Ferguson JR, Symington LS . The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol Cell Biol 1999; 19:556–566.
Article CAS PubMed PubMed Central Google Scholar - Ivanov EL, Sugawara N, White CI, Fabre F, Haber JE . Mutations in XRS2 and RAD50 delay but do not prevent mating-type switching in Saccharomyces cerevisiae. Mol Cell Biol 1994; 14:3414–3425.
Article CAS PubMed PubMed Central Google Scholar - Milne GT, Jin S, Shannon KB, Weaver DT . Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol Cell Biol 1996; 16:4189–4198.
Article CAS PubMed PubMed Central Google Scholar - Moore JK, Haber JE . Cell-cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol 1996; 16:2164–2173.
Article CAS PubMed PubMed Central Google Scholar - Kramer KM, Brock JA, Bloom K, Moore JK, Haber JE . Two different types of double-strand breaks in Saccharomyces cerevisiae are repaired by similar _RAD52_-independent, nonhomologous recombination events. Mol Cell Biol 1994; 14:1293–1301.
Article CAS PubMed PubMed Central Google Scholar - Hammarsten O, DeFazio LG, Chu G . Activation of DNA-dependent protein kinase by single-stranded DNA ends. J Biol Chem 2000; 275:1541–1550.
Article CAS PubMed Google Scholar - Budman J, Chu G . Processing of DNA for nonhomologous end-joining by cell-free extract. EMBO J 2005; 24:849–860.
Article CAS PubMed PubMed Central Google Scholar - Boulton SJ . Cellular functions of the BRCA tumour-suppressor proteins. Biochem Soc Trans 2006; 34:633–645.
Article CAS PubMed Google Scholar - Brenneman MA . BRCA1 and BRCA2 in DNA repair and genome stability. In: Nickoloff JA, Hoekstra MF, eds. DNA Damage and Repair, Vol 3: Advances from Phage to Humans. Totowa, NJ: Humana Press, 2001:237–267.
- Jung D, Giallourakis C, Mostoslavsky R, Alt FW . Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol 2006; 24:541–570.
Article CAS PubMed Google Scholar - Cremer T, Cremer M, Dietzel S, et al. Chromosome territories – a functional nuclear landscape. Curr Opin Cell Biol 2006; 18:307–316.
Article CAS PubMed Google Scholar - Dekker J, Rippe K, Dekker M, Kleckner N . Capturing chromosome conformation. Science 2002; 295:1306–1311.
Article CAS PubMed Google Scholar - Mortimer RK . Radiobiological and genetic studies on a polyploid series (haploid to hexaploid) of Saccharomyces cerevisiae. Radiat Res 1958; 9:312–326.
Article CAS PubMed Google Scholar - Astrom SU, Okamura SM, Rine J . Yeast cell-type regulation of DNA repair. Nature 1999; 397:310.
Article CAS PubMed Google Scholar - Lee SE, Paques F, Sylvan J, Haber JE . Role of yeast SIR genes and mating type in directing DNA double-strand breaks to homologous and non-homologous repair paths. Curr Biol 1999; 9:767–770.
Article CAS PubMed Google Scholar - Nickoloff JA, Haber JE . Mating-type control of DNA repair and recombination in Saccharomyces cerevisiae. In: Nickoloff JA, Hoekstra MF, eds. DNA Damage and Repair, Vol 3: Advances from Phage to Humans. Totowa, NJ: Humana Press, 2001:107–124.
- Allen C, Kurimasa A, Brennemann MA, Chen DJ, Nickoloff JA . DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination. Proc Natl Acad Sci USA 2002; 99:3758–3763.
Article CAS PubMed PubMed Central Google Scholar - Pierce AJ, Hu P, Han MG, Ellis N, Jasin M . Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev 2001; 15:3237–3242.
Article CAS PubMed PubMed Central Google Scholar - Delacote F, Han M, Stamato TD, Jasin M, Lopez BS . An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res 2002; 30:3454–3463.
Article CAS PubMed PubMed Central Google Scholar - Ooi SL, Shoemaker DD, Boeke JD . A DNA microarray-based genetic screen for nonhomologous end-joining mutants in Saccharomyces cerevisiae. Science 2001; 294:2552–2556.
Article CAS PubMed Google Scholar - Valencia M, Bentele M, Vaze MB, et al. NEJ1 controls non-homologous end joining in Saccharomyces cerevisiae. Nature 2001; 414:666–669.
Article CAS PubMed Google Scholar - Kegel A, Sjostrand JO, Astrom SU . Nej1p, a cell type-specific regulator of nonhomologous end joining in yeast. Curr Biol 2001; 11:1611–1617.
Article CAS PubMed Google Scholar - Valencia-Burton M, Oki M, Johnson J, et al. Different mating-type-regulated genes affect the DNA repair defects of Saccharomyces RAD51, RAD52 and RAD55 mutants. Genetics 2006; 174:41–55.
Article CAS PubMed PubMed Central Google Scholar - Yamazoe M, Sonoda E, Hochegger H, Takeda S . Reverse genetic studies of the DNA damage response in the chicken B lymphocyte line DT40. DNA Repair (Amst) 2004; 3:1175–1185.
Article CAS Google Scholar - Aladjem MI, Itoh N, Utiyama H, Wahl GM . ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr Biol 1998; 8:145–155.
Article CAS PubMed Google Scholar - Mekeel KL, Tang W, Kachnic LA, et al. Inactivation of p53 results in high-rates of homologous recombination. Oncogene 1997; 14:1847–1857.
Article CAS PubMed Google Scholar - Willers H, McCarthy EE, Wu B, et al. Dissociation of p53-mediated suppression of homologous recombination from G1/S cell cycle checkpoint control. Oncogene 2000; 19:632–639.
Article CAS PubMed Google Scholar - Romanova LY, Willers H, Blagosklonny MV, Powell SN . The interaction of p53 with replication protein A mediates suppression of homologous recombination. Oncogene 2004; 23:9025–9033.
Article CAS PubMed Google Scholar - Akyuz N, Boehden GS, Susse S, et al. DNA substrate dependence of p53-mediated regulation of double-strand break repair. Mol Cell Biol 2002; 22:6306–6317.
Article CAS PubMed PubMed Central Google Scholar - Gatz SA, Wiesmuller L . p53 in recombination and repair. Cell Death Differ 2006; 13:1003–1016.
Article CAS PubMed Google Scholar - Yoon D, Wang Y, Stapleford K, Wiesmuller L, Chen J . P53 inhibits strand exchange and replication fork regression promoted by human Rad51. J Mol Biol 2004; 336:639–654.
Article CAS PubMed Google Scholar - Orii KE, Lee Y, Kondo N, McKinnon PJ . Selective utilization of nonhomologous end-joining and homologous recombination DNA repair pathways during nervous system development. Proc Natl Acad Sci USA 2006; 103:10017–10022.
Article CAS PubMed PubMed Central Google Scholar - Kadyk LC, Hartwell LH . Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 1992; 132:387–402.
CAS PubMed PubMed Central Google Scholar - Watrin E, Peters JM . Cohesin and DNA damage repair. Exp Cell Res 2006; 312:2687–2693.
Article CAS PubMed Google Scholar - Takata M, Sasaki MS, Sonoda E, et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 1998; 17:5497–5508.
Article CAS PubMed PubMed Central Google Scholar - Chen FQ, Nastasi A, Shen ZY, et al. Cell cycle-dependent protein expression of mammalian homologs of yeast DNA double-strand break repair genes RAD51 and RAD52. Mutat Res 1997; 384:205–211.
Article CAS PubMed Google Scholar - Toone WM, Aerne BL, Morgan BA, Johnston LH . Getting started: regulating the initiation of DNA replication in yeast. Annu Rev Microbiol 1997; 51:125–149.
Article CAS PubMed Google Scholar - Ira G, Pellicioli A, Balijja A, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004; 431:1011–1017.
Article CAS PubMed PubMed Central Google Scholar - Aylon Y, Liefshitz B, Kupiec M . The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J 2004; 23:4868–4875.
Article CAS PubMed PubMed Central Google Scholar - Ferreira MG, Cooper JP . The fission yeast Taz1 protein protects chromosomes from Ku-dependent end-to-end fusions. Mol Cell 2001; 7:55–63.
Article CAS PubMed Google Scholar - Caspari T, Murray JM, Carr AM . Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev 2002; 16:1195–1208.
Article CAS PubMed PubMed Central Google Scholar - Esashi F, Christ N, Gannon J, et al. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005; 434:598–604.
Article CAS PubMed Google Scholar - Esashi F, Galkin VE, Yu X, Egelman EH, West SC . Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat Struct Mol Biol 2007; 14:468–474.
Article CAS PubMed Google Scholar - Douglas P, Sapkota GP, Morrice N, et al. Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA-dependent protein kinase. Biochem J 2002; 368:243–251.
Article CAS PubMed PubMed Central Google Scholar - Chan DW, Chen BP-C, Prithivirasingh S, et al. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev 2002; 16:2333–2338.
Article CAS PubMed PubMed Central Google Scholar - Chen BP, Chan DW, Kobayashi J, et al. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem 2005; 280:14709–14715.
Article CAS PubMed Google Scholar - Douglas P, Cui X, Block WD, et al. The DNA-dependent protein kinase catalytic subunit is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein kinase domain. Mol Cell Biol 2007; 27:1581–1591.
Article CAS PubMed Google Scholar - Kurimasa A, Kumano S, Boubnov NV, et al. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol Cell Biol 1999; 19:3877–3884.
Article CAS PubMed PubMed Central Google Scholar - Ding Q, Reddy YV, Wang W, et al. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol Cell Biol 2003; 23:5836–5848.
Article CAS PubMed PubMed Central Google Scholar - Lee SE, Mitchell RA, Cheng A, Hendrickson EA . Evidence for DNA-PK-dependent and DNA-PK-independent DNA double-strand break repair pathways in mammalian cells as a function of the cell cycle. Mol Cell Biol 1997; 17:1425–1433.
Article CAS PubMed PubMed Central Google Scholar - Roth D, Wilson JH . Relative rates of homologous and nonhomologous recombination in transfected DNA. Proc Natl Acad Sci USA 1985; 82:3355–3359.
Article CAS PubMed PubMed Central Google Scholar - Haber JE . Partners and pathways: repairing a double-strand break. Trends Biochem Sci 2000; 16:259–264.
CAS Google Scholar - Baumann P, West SC . Role of the human RAD51 protein in homologous recombination and double-stranded break repair. Trends Biochem Sci 1998; 23:247–251.
Article CAS PubMed Google Scholar - Haber JE . The many interfaces of Mre11. Cell 1998; 95:583–586.
Article CAS PubMed Google Scholar - Kim JS, Krasieva TB, Kurumizaka H, et al. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J Cell Biol 2005; 170:341–347.
Article CAS PubMed PubMed Central Google Scholar - Chen JJ, Silver D, Cantor S, Livingston DM, Scully R . BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer Res 1999; 59:S1752–S1756.
Google Scholar - Chen J, Silver DP, Walpita D, et al. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cell 1998; 2:317–328.
Article CAS PubMed Google Scholar - Gudmundsdottir K, Ashworth A . The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006; 25:5864–5874.
Article CAS PubMed Google Scholar - Paull TT, Cortez D, Bowers B, Elledge SJ, Gellert M . Direct DNA binding by Brca1. Proc Natl Acad Sci USA 2001; 98:6086–6091.
Article CAS PubMed PubMed Central Google Scholar - Zhuang J, Zhang J, Willers H, et al. Checkpoint kinase 2-mediated phosphorylation of BRCA1 regulates the fidelity of nonhomologous end-joining. Cancer Res 2006; 66:1401–1408.
Article CAS PubMed Google Scholar - Downey M, Durocher D . γH2AX as a checkpoint maintenance signal. Cell Cycle 2006; 5:1376–1381.
Article CAS PubMed Google Scholar - Downs JA, Lowndes NF, Jackson SP . A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 2000; 408:1001–1004.
Article CAS PubMed Google Scholar - Moore JD, Yazgan O, Ataian Y, Krebs JE . Diverse roles for histone H2A modifications in DNA damage response pathways in yeast. Genetics 2007; 176:15–25.
Article CAS PubMed PubMed Central Google Scholar - Xie A, Puget N, Shim I, et al. Control of sister chromatid recombination by histone H2AX. Mol Cell 2004; 16:1017–1025.
Article CAS PubMed Google Scholar - Wu S, Shi Y, Mulligan P, et al. A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nat Struct Mol Biol 2007 Nov 18; doi:10.1038/nsmb1332.
Article CAS Google Scholar - Osley MA, Tsukuda T, Nickoloff JA . ATP-dependent chromatin remodeling factors and DNA damage repair. Mutat Res 2007; 618:65–80.
Article CAS PubMed PubMed Central Google Scholar - Downs JA, Nussenzweig MC, Nussenzweig A . Chromatin dynamics and the preservation of genetic information. Nature 2007; 447:951–958.
Article CAS PubMed Google Scholar - Saberi A, Hochegger H, Szuts D, et al. RAD18 and poly(ADP-ribose) polymerase independently suppress the access of nonhomologous end joining to double-strand breaks and facilitate homologous recombination-mediated repair. Mol Cell Biol 2007; 27:2562–2571.
Article CAS PubMed PubMed Central Google Scholar - Dominguez-Bendala J, Masutani M, McWhir J . Down-regulation of PARP-1, but not of Ku80 or DNA-PKcs', results in higher gene targeting efficiency. Cell Biol Int 2006; 30:389–393.
Article CAS PubMed Google Scholar - Allen C, Halbrook J, Nickoloff JA . Interactive competition between homologous recombination and non-homologous end joining. Mol Cancer Res 2003; 1:913–920.
CAS PubMed Google Scholar - Chan DW, Lees-Miller SP . The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J Biol Chem 1996; 271:8936–8941.
Article CAS PubMed Google Scholar - Calsou P, Frit P, Humbert O, et al. The DNA-dependent protein kinase catalytic activity regulates DNA end processing by means of Ku entry into DNA. J Biol Chem 1999; 274:7848–7856.
Article CAS PubMed Google Scholar - Merkle D, Douglas P, Moorhead GBG, et al. The DNA-dependent protein kinase interacts with DNA to form a protein-DNA complex that is disrupted by phosphorylation. Biochemistry 2002; 41:12706–12714.
Article CAS PubMed Google Scholar - Douglas P, Moorhead GBG, Ye RQ, Lees-Miller SP . Protein phosphatases regulate DNA-dependent protein kinase activity. J Biol Chem 2001; 276:18992–18998.
Article CAS PubMed Google Scholar - Block WD, Yu Y, Merkle D, et al. Autophosphorylation-dependent remodeling of the DNA-dependent protein kinase catalytic subunit regulates ligation of DNA ends. Nucleic Acids Res 2004; 32:4351–4357.
Article CAS PubMed PubMed Central Google Scholar - Reddy YV, Ding Q, Lees-Miller SP, Meek K, Ramsden DA . Non-homologous end joining requires that the DNA-PK complex undergo an autophosphorylation-dependent rearrangement at DNA ends. J Biol Chem 2004; 279:39408–39413.
Article CAS PubMed Google Scholar - Convery E, Shin EK, Ding Q, et al. Inhibition of homologous recombination by variants of the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs). Proc Natl Acad Sci USA 2005; 102:1345–1350.
Article CAS PubMed PubMed Central Google Scholar - Peng Y, Woods RG, Beamish H, et al. Deficiency in the catalytic subunit of DNA-dependent protein kinase causes down-regulation of ATM. Cancer Res 2005; 65:1670–1677.
Article CAS PubMed Google Scholar - Chen BP, Uematsu N, Kobayashi J, et al. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem 2007; 282:6582–6587.
Article CAS PubMed Google Scholar - Meyn MS . High spontaneous intrachromosomal recombination rates in ataxia-telangiectasia. Science 1993; 260:1327–1330.
Article CAS PubMed Google Scholar - Morrison C, Sonoda E, Takao N, et al. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J 2000; 19:463–471.
Article CAS PubMed PubMed Central Google Scholar - Luo C-M, Tang W, Mekeel KL, et al. High frequency and error-prone DNA recombination in ataxia telangiectasia cell lines. J Biol Chem 1996; 271:4497–4503.
Article CAS PubMed Google Scholar - Bolderson E, Scorah J, Helleday T, Smythe C, Meuth M . ATM is required for the cellular response to thymidine induced replication fork stress. Hum Mol Genet 2004; 13:2937–2945.
Article CAS PubMed Google Scholar - Sakamoto S, Iijima K, Mochizuki D, et al. Homologous recombination repair is regulated by domains at the N- and C-terminus of NBS1 and is dissociated with ATM functions. Oncogene 2007; 26:6002–6009.
Article CAS PubMed Google Scholar - Barchi M, Mahadevaiah S, Di Giacomo M, et al. Surveillance of different recombination defects in mouse spermatocytes yields distinct responses despite elimination at an identical developmental stage. Mol Cell Biol 2005; 25:7203–7215.
Article CAS PubMed PubMed Central Google Scholar - Wang H, Boecker W, Wang H, et al. Caffeine inhibits homology directed repair of I-SceI induced DNA double-strand breaks. Oncogene 2004; 23:824–834.
Article CAS PubMed Google Scholar - Hickson I, Zhao Y, Richardson CJ, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 2004; 64:9152–9159.
Article CAS PubMed Google Scholar - Lundin C, Erixon K, Arnaudeau C, et al. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol Cell Biol 2002; 22:5869–5878.
Article CAS PubMed PubMed Central Google Scholar - Kuhne M, Riballo E, Rief N, et al. A double-strand break repair defect in ATM-deficient cells contributes to radiosensitivity. Cancer Res 2004; 64:500–508.
Article PubMed Google Scholar - Bredemeyer AL, Sharma GG, Huang CY, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 2006; 442:466–470.
Article CAS PubMed Google Scholar
Acknowledgements
We thank D Chen and BPC Chen (University of Texas Southwestern Medical Center, USA), S Lees-Miller (University of Calgary, Canada), K Meek (Michigan State University, USA), MA Osley (University of New Mexico, USA), and members of the Nickoloff lab for helpful discussions and excellent collaborations. Our research on the NHEJ/HR interface is supported by National Cancer Institute Grant R01 CA100862 to JA Nickoloff.
Author information
Authors and Affiliations
- Department of Molecular Genetics and Microbiology, University of New Mexico School of Medicine and Cancer Center, Albuquerque, 87131, NM, USA
Meena Shrivastav, Leyma P De Haro & Jac A Nickoloff
Authors
- Meena Shrivastav
You can also search for this author inPubMed Google Scholar - Leyma P De Haro
You can also search for this author inPubMed Google Scholar - Jac A Nickoloff
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toJac A Nickoloff.
Rights and permissions
About this article
Cite this article
Shrivastav, M., De Haro, L. & Nickoloff, J. Regulation of DNA double-strand break repair pathway choice.Cell Res 18, 134–147 (2008). https://doi.org/10.1038/cr.2007.111
- Published: 24 December 2007
- Issue Date: January 2008
- DOI: https://doi.org/10.1038/cr.2007.111