Perceiving the epigenetic landscape through histone readers (original) (raw)
. Author manuscript; available in PMC: 2013 May 6.
Published in final edited form as: Nat Struct Mol Biol. 2012 Dec;19(12):1218–1227. doi: 10.1038/nsmb.2436
Abstract
Post-translational modifications (PTMs) of histones provide a fine-tuned mechanism for regulating chromatin structure and dynamics. PTMs can alter direct interactions between histones and DNA and serve as docking sites for protein effectors, or readers, of these PTMs. Binding of the readers recruits or stabilizes various components of the nuclear signaling machinery at specific genomic sites, mediating fundamental DNA-templated processes, including gene transcription and DNA recombination, replication and repair. In this review, we highlight the latest advances in characterizing histone-binding mechanisms and identifying new epigenetic readers and summarize the functional significance of PTM recognition.
The genetic material of eukaryotic cells is packaged into the nucleus in the form of chromatin. Chromatin is made up of building blocks called nucleosomes. Each nucleosomal particle contains an octamer of four histone proteins—H2A, H2B, H3 and H4—around which genomic DNA is wound almost twice1. The nucleosomes undergo recurrent structural rearrangements through DNA unwrapping and rewrapping and histone core disassembly and assembly, and they are subject to covalent modifications. The modifications, or epigenetic marks, have been identified on both DNA and histones. Whereas DNA can primarily be methylated, histones are capable of carrying a wide array of PTMs2. A particularly large number of PTMs have been discovered on the histone tails that protrude from the nucleosomal core and are freely accessible to enzymes for the deposition or removal of PTMs (Fig. 1). The mechanisms by which histone PTMs affect chromatin structure and dynamics can generally be divided into two categories. Histone PTMs can directly influence histone-DNA and histone-histone interactions, or they can be targeted by protein effectors (also referred to as histone-binding domains or readers of PTMs).
Figure 1.

Readers of histone PTMs. Recognition of the methylated (me) lysine, methylated (me) arginine, acetylated (ac) lysine and phosphorylated (ph) serine and threonine residues of the N-terminal histone H3 tail by indicated readers.
The specific recognition of PTMs by readers recruits various components of the nuclear signaling network to chromatin, mediating fundamental processes such as gene transcription, DNA replication and recombination, DNA damage response and chromatin remodeling. Chromatin-associating complexes often contain multiple readers within one or several subunits that show specificities for distinct PTMs. Coordinated binding to multiple PTMs can provide a lock-and- key–type mechanism for targeting particular genomic sites and ensuring the proper biological outcomes. Misreading of epigenetic marks has been shown to underlie a host of human diseases, including autoimmune and developmental abnormalities and cancer3,4. More recently, misregulation of epigenetic pathways has been implicated in addiction, schizophrenia and other mental disorders5,6. Thus, understanding the molecular mechanism and functional significance of reader-PTM interactions is essential to understanding not only the basic mechanisms of epigenetic regulation but also the etiology of epimutation-induced human diseases.
In this review, we outline known readers of histone PTMs, detail their mechanisms of action and discuss cross-talk between protein effectors and consequences of the combinatorial readout of PTMs.
History of histone PTMs
The first PTMs of histones were discovered nearly 50 years ago7; however, it was not until 30 years later that enzymatic activities of a histone acetyltransferase (HAT) and a deacetylase were directly linked to transcriptional regulation8,9. In addition to lysine acetylation, numerous PTMs have now been identified, including methylation, ubiquitination, SUMOylation, crotonylation, butyrylation and propionylation of lysine residues; methylation, citrullination and ADP-ribosylation of arginine residues; and phosphorylation and glycosylation of serine and threonine residues. As the substantial degree of interplay between PTMs began to unfold, it led Strahl and Allis to propose the histone code hypothesis, which states that “multiple histone modifications, acting in a combinatorial or sequential fashion on one or multiple histone tails, specify unique downstream function”10. Soon afterward, the terms ‘writer’, ‘eraser’ and ‘reader’ were formulated to describe proteins that deposit, remove and recognize PTMs, respectively11,12. In addition to histone PTMs, other factors—including methylation and hydroxymethylation of DNA, histone variants, nucleosome positioning, noncoding RNAs and histone chaperones—are necessary for fine-tuning chromatin structure and function, and together they constitute the powerful and dynamic epigenetic machinery.
The term ‘epigenetics’ was originally associated with heritable changes in gene activity that occur without alterations in the genetic code, sometimes defined as ‘soft’ inheritance13,14. However, more and more often this term is used to describe DNA-related regulatory mechanisms that do not involve changes in the nucleotide sequence, regardless of whether such imprinting is strictly heritable (‘epi-’ is derived from the Greek for ‘above’; hence, ‘above genetics’). The intricate relationship among the epigenetic elements represents one of the most intriguing concepts in modern chromatin biology, which we have only begun to explore. Clearly, the spatial and temporal modulation of, and cross-talk between, histone PTMs has a very important role in defining the chromatin landscape.
The first reader of histone PTMs was discovered in 1999, when the bromodomain of the HAT PCAF was found to recognize acetylated lysine15. It was also the first example supporting the hypothesis that bromodomains contribute to “acetylation by tethering transcriptional HATs to specific chromosomal sites”15. Since then, a large number of histone effectors have been identified and characterized, including readers of methylated lysine and arginine and phosphorylated serine and threonine16. Moreover, many readers can distinguish a particular sequence surrounding a PTM, affording specific chromatin targeting ability to their host proteins. Here we outline the known protein effectors and the molecular mechanisms of their interaction with target PTMs.
Methylation readers
Methylation is perhaps the most versatile of all histone PTMs. Two residues—lysine and arginine—can be methylated, and each has three possible methylation states. Unlike other modifications, methylation does not change the overall charge, although it does alter the hydrophobic character and size of the modified residue.
Lysine methylation
Lysine is methylated on its ε-amino group and can be mono-, di- and trimethylated. The canonical sites for methylation comprise six lysine residues of histone H3 (K4, K9, K26, K27, K36 and K79), K20 of histone H4 and K26 of histone H1. With the exception of H3K79, these are all located in the N-terminal tails of the histone proteins. To date, readers of methyllysine are the most thoroughly characterized group and include ADD (ATRX-DNMT3-DNMT3L), ankyrin, bromo-adjacent homology (BAH), chromobarrel, chromodomain, double chromodomain (DCD), MBT (malignant brain tumor), PHD (plant homeodomain), PWWP (Pro-Trp-Trp-Pro), tandem Tudor domain (TTD), Tudor, WD40 and the zinc finger CW (zf-CW) (Table 1 and Fig. 2a).
Table 1.
Histone readers and their target PTMs
| Recognition of | Reader | Histone PTM |
|---|---|---|
| Methyllysine | ADD | H3K9me3 |
| Ankyrin | H3K9me2, H3K9me1 | |
| BAH | H4K20me2 | |
| Chromobarrel | H3K36me3, H3K36me2, H4K20me1, H3K4me1 | |
| Chromodomain | H3K9me3, H3K9me2, H3K27me3, H3K27me2 | |
| DCD | H3K4me3, H3K4me2, H3K4me1 | |
| MBT | H3Kme1, H3Kme2, H4Kme1, H4Kme2 | |
| PHD | H3K4me3, H3K4me2, H3K9me3 | |
| PWWP | H3K36me3, H4K20me1, H4K20me3, H3K79me3 | |
| TTD | H3K4me3, H3K9me3, H4K20me2 | |
| Tudor | H3K36me3 | |
| WD40 | H3K27me3, H3K9me3 | |
| zf-CW | H3K4me3 | |
| Methylarginine | ADD | H4R3me2s |
| Tudor | H3Rme2, H4Rme2 | |
| WD40 | H3R2me2 | |
| Acetyllysine | Bromodomain | H3Kac, H4Kac, H2AKac, H2BKac |
| DBD | H3KacKac, H4KacKac | |
| DPF | H3Kac | |
| Double PH | H3K56ac | |
| Phosphoserine or phosphothreonine | 14-3-3 | H3S10ph, H3S28ph |
| BIR | H3T3ph | |
| Tandem BRCT | H2AXS139ph | |
| Unmodified histone | ADD | H3un |
| PHD | H3un | |
| WD40 | H3un |
Figure 2.

Molecular mechanisms for the recognition of methyllysine and methylarginine. (a) Structures of the readers in complex with histone peptides methylated at lysine residues. The aromatic cage residues and the histone peptides are colored blue and red, respectively. (b–d) The binding sites for trimethylated lysine (Kme3) (b), dimethylated lysine (Kme2) (c) and symmetrically dimethylated arginine (Rme2s) (d). PDB codes: 3B95, 4DOW, 1KNE, 2PQW, 2G6Q, 2X4X, 2IG0, 3IIW, 2F6J, 4A7J and 4A4E.
The foremost trait of the methyllysine-specific readers is that they bind this PTM through an aromatic cage, typically formed by two to four aromatic residues (Fig. 2b). In many complexes the aromatic rings are positioned perpendicular to each other, surrounding the fully extended side chain of the methylated lysine. The complex formation is driven by cation-π interactions between the methylammonium group and the aromatic rings as well as hydrophobic and van der Waals contacts. The mono-, di- or trimethylated state of lysine is selected for by the exact composition and size of the pocket. A reader prefers mono- or dimethylated lysine over trimethylated lysine if one of the walls of the cage is replaced by a negatively charged aspartate or glutamate residue, the carboxylate group of which makes additional favorable hydrogen bonding and electrostatic contacts with the methylammonium moiety (Fig. 2c). A small pocket size can also preclude interaction with a higher methylation state owing to steric hindrance, whereas a larger pocket selects for a higher methylation state, as the necessary contacts are possible only with the bulkier methylammonium group.
Specificity for a particular methylated lysine is imparted by interaction with surrounding residues. Some histone readers show high degrees of specificity, whereas others are selective for only a certain methylation state and otherwise bind very promiscuously. Beyond caging of the methyllysine, the mechanism of recognition of surrounding residues varies among readers.
The structurally related chromodomain, chromobarrel, MBT, PWWP, Tudor and TTD modules possess a characteristic β-barrel topology and comprise the Royal superfamily. The chromodomain is the smallest member, consisting of four curved β-strands and an α-helix. The chromodomains of HP1 and Polycomb were found to recognize histone H3 trimethylated at K9 (H3K9me3) and H3K27me3, respectively, and these proteins were the first examples of readers specific for methyllysine17–21. Chromodomains generally prefer trimethylated lysine, though some have been shown to bind dimethylated species. The aromatic cage of the chromodomain of mouse and fly HP1 contains an aspartate or glutamate residue, accounting for its ability to interact well with H3K9me3 and dimethylated H3K9 (H3K9me2) (refs. 18,19). Upon binding, the histone peptide adopts a β-strand conformation and inserts between two β-strands of the chromodomain, completing the five-stranded antiparallel β-barrel. This induced-fit binding mode is stabilized through backbone hydrogen bonds and electrostatic contacts involving up to seven residues preceding trimethylated lysine and one residue following trimethylated lysine of the histone peptide. A small hydrophobic pocket, in which the _n_–2 (with respect to methyllysine) residue resides, precludes binding of H3K4me3, whereas interactions at the _n_–4 and _n_–5 positions are important for distinguishing between H3K9me3 and H3K27me3 (ref. 22).
Other Royal superfamily members have a complete five-stranded β-barrel, which prevents the insertion of a peptide between β-strands of the barrel. Instead, the histone tail lies across an open edge of the β-barrel, with the methyllysine occupying the aromatic cage located near the upper rim. Tudor is the classic module that utilizes this binding mechanism. Tudor can exist as a single domain or in tandem, containing two β-barrels. The single Tudor domains of PHF1 and PHF19 have been shown to recognize H3K36me3, whereas the canonical TTD of 53BP1 associates with H4K20me2, and the hybrid TTD of JMJD2A binds H3K4me3 and H4K20me3 (refs. 23–27,153). The aromatic cage is generally seen in only one β-barrel of TTDs, and the majority of contacts with the histone tail are found within that barrel. A notable exception to this is the TTD of Sgf29 (ref. 28). In the complex of the Sgf29 TTD with H3K4me3, the peptide spans both Tudor domains, with A1 and K4me3 being bound in separate β-barrels28. Here, the specificity toward H3K4 is largely defined by A1 recognition, a common determinant of H3K4me3 binding.
MBT reads lower methylation states, recognizing mono- and dimethylated lysines. All MBTs characterized at present contain two to four repeats of ~100 amino acids. The most common three-repeat 3MBT module folds in a propeller-like structure, and the four-repeat 4MBT module has an asymmetric rhombus architecture29–32. Notably, although each MBT repeat contains an aromatic cage, which also includes an acidic residue, only the second repeat binds methyllysine29–32. Very little sequence specificity is observed, as few contacts are made with the histone residues beyond mono- or dimethylated lysine (refs. 29–33). The other two members of the Royal family, PWWP and chromobarrel, show some specificity, preferring tri- or dimethylated H3K36 and monomethylated H4K20 (H4K20me1); however, they bind rather weakly34–39.
Distinct from the Royal family is the PHD finger, a well-characterized reader of H3K4me3 (refs. 40–43). It contains a C4HC3 motif that coordinates two zinc ions in a cross-brace manner. Such an arrangement produces a globular domain with a small β-sheet and an α-helix. PHD fingers make extensive contacts with H3K4me3, imparting a high degree of specificity44. The peptide forms the third antiparallel β-strand, pairing with the existing double-stranded β-sheet of the protein. While K4me3 inserts into the aromatic cage, R2 occupies the adjacent binding pocket. This pocket often contains acidic residues, which restrain the guanidinium group of R2 through ionic and hydrogen-bonding interactions. A1 is bound in a small hydrophobic cavity, and its N-terminal amino group donates two or three hydrogen bonds to the backbone carbonyl groups of the protein. This strictly conserved coordination of A1 and a frequent constraining of R2, as well as the fitting of T3 into a small pocket, provide the specificity for K4me3. A similar mode of recognition of the histone A1R2T3K4me3 sequence is seen in a single zinc finger (zf-CW)45 and in the DCD of human CHD1, in which two chromodomains fold in a unique module, forming a conjoint binding site for the peptide46.
The WD40 domain of EED binds trimethylated lysines through an aromatic cage positioned at the top of the channel of a seven-bladed β-propeller47,48. It is fairly promiscuous and interacts with H3K27me3, H3K9me3, H4K20me3 and H1K26me3. The structure of the EED WD40 in complex with H3K27me3 reveals an important role for the residues flanking K27me3 (refs. 47,48). A small hydrophobic residue, a solvent-exposed residue and another hydrophobic residue at positions −2, minus;1 and +2, respectively, are responsible for the preferential recognition of repressive chromatin marks and the disfavoring of H3K4me3. Owing to extensive intermolecular contacts, the ankyrin repeats of G9a and GLP and the BAH domain of ORC1 show high specificity for di- or monomethylated H3K9 and H4K20me2, respectively49,50. The H3K9me2 peptide is sandwiched between the β-turns and α-helices of the fourth and fifth ankyrin repeats, whereas K9me2 fits into the aromatic cage also containing a glutamate. Residues S10–G13 of the H3K9me2 peptide are involved in multiple interactions that impart specificity. The ATRX ADD domain and the PHD fingers of CHD4 and TRIM33 are examples of readers lacking the aromatic cage51–55. Binding of these modules to the histone H3 tail is enhanced by methylation of H3K9. K9me3 is uniquely coordinated through formation of a nonconventional carbon-oxygen hydrogen bond or hydrophobic and cation-π contacts with a single aromatic residue.
Functional significance of Kme recognition
Methylation of lysine residues is the most characterized PTM in terms of cellular functions, hallmarked by its major role in transcriptional regulation. On a global chromatin level, H3K4me is considered to be a gene-activation mark; however, the consequence of its readout by histone effectors is highly context dependent. Lysine trimethylation is generally located in the 5′ end region of actively transcribed genes, and this trait is conserved from yeast to higher eukaryotes56–58. The TAF3 subunit of the basal transcription complex TFIID binds to H3K4me3 through its PHD finger, directly implicating this mark in transcriptional activation59. In contrast, recognition of H3K4me3 by the PHD finger of ING2, a subunit of the mSin3a histone deacetylase complex, links this PTM to rapid gene repression42. Binding of the PHD finger of the PHF8 histone demethylase60 and the DCD of the CHD1 ATPase46 to H3K4me3 mediates enzymatic activities of these proteins, coupling the readout of H3K4me to the chromatin-modifying and chromatin-remodeling mechanisms, respectively. The chromo-barrel domain of the Tip60 acetyltransferase associates with H3K4me1, a marker of active and poised enhancer elements, tethering Tip60 to the enhancer regions and leading to estrogen-induced transcription61.
H3K36me also has a role in the transcriptional process. Genome-wide profiling of H3K36me stages shows a progressive shift from monomethylation to trimethylation of K36 between the 5′ and 3 ′ ends of genes62. The H3K36me-specific enzyme Set2 binds to elongating RNA polymerase II and methylates the coding regions of genes, preventing spurious transcription initiation and histone exchange in budding yeast by recruiting the Rpd3S deacetylase complex via its Eaf3 subunit, which binds to K36me through its chromobarrel domain63–65. This complex deacetylates nucleosomes on gene bodies during elongation and thus provides nucleosome stability in the wake of elongating RNA polymerase II66. Recognition of H3K36me3 by the Tudor domain of PHF1 inhibits Polycomb repressive complex 2 (PRC2)-mediated H3K27 methylation27. This interaction may represent a mechanism to guide the deposition of H3K27me3, restricting activity of the PRC2 complex at the boundaries of active and repressed regions and preventing the Polycomb-linked repressive environment from spreading into neighboring transcriptionally active chromatin67,68. Interaction of another component of the PRC2 complex, PHF19, with H3K36me3 is important for localization of PHF19 at a subset of PRC2 target genes and is required for their repression and H3K27me3 deposition in mouse embryonic stem cells23, as well as for the recruitment of demethylase NO66 to embryonic stem cell genes during differentiation (ref. 153).
The importance of H3K36me recognition for other fundamental processes has recently been established. Association of the chromo-barrel domains of MRG15 and the MSL3 subunit of the MOF complex with H3K36me is essential in regulating alternative splicing of mRNAs69 and upregulating genes for dosage compensation in Drosophila melanogaster70, respectively. The PWWP domain present in the IOC4 subunit of the ISW1b chromatin-remodeling complex also recognizes H3K36me71,72, bridging readout of this PTM with ATP-dependent nucleosome remodeling activity. An additional role for H3K36me in DNA repair and recombination through the LEDGF PWWP domain has been suggested73; however, it remains unclear whether this PTM has a positive or negative regulatory function in DNA replication74,75. Recently, the BAH domain of ORC1 has been shown to bind H4K20me2, and this interaction aids in DNA replication licensing50. The H4K20me2 mark also has a crucial role in the DNA repair pathway in which it is targeted by the TTD of 53BP1 (ref. 24). This interaction is in direct competition with the JMJD2A and JMJD2B histone demethylases, which bind to H4K20me2 through their TTDs before DNA damage76. Upon DNA damage, JMJD2 is degraded through the RNF8- and RNF168-dependent pathways, exposing H4K20me2 to the 53BP1 TTD76.
Recognition of the H3K9 and H3K27 methylation marks is largely associated with formation of constitutive and facultative heterochromatin and gene silencing. H3K27me1 and H3K9me3 are found in pericentromeric heterochromatin regions77,78, whereas H3K27me3 and H3K9me2 colocalize in repressed euchromatin regions. The K27me and K9me marks are both targets for chromodomain-containing proteins. The former recruits Polycomb proteins and their host PRC1 complexes to repressed chromatin20,21, whereas the latter recruits HP1 proteins involved in heterochromatin formation and spreading17,79,80.
Arginine methylation
The omega nitrogen atoms of arginine can be monomethylated or dimethylated symmetrically (Rme2s) or asymmetrically (Rme2a). To date, methylation of H3R2, H3R8, H3R17, H3R26, H4R3, H2AR11 and H2AR29 has been reported. As compared to lysine methylation, information on writing and especially erasing and recognition of methylated arginine is rather limited; however, there are some indications that this mark is read by histone effectors, and a number of readers show sensitivity to the methylation state of arginine.
The Tudor domain is known to bind methylated arginines in nonhistone ligands, particularly PIWI81, and recent studies suggest that the TDRD3 Tudor domain recognizes H3R17me2a and H4R3me2a82,83. Although no structure has yet been determined of a Tudor bound to a histone peptide containing a methylated arginine, the structures of the SMN and SPF30 Tudor domains in complex with a dimethylated arginine amino acid provide insight into how this mark can be specifically recognized84. Like methyllysine, methylated arginine occupies an aromatic cage at the top of the Tudor β-barrel (Fig. 2d). However, this cage is much narrower than the cage for methyllysine and thus favors the planar guanidinium group.
The WD40 repeat of WDR5 prefers H3R2me2s over H3 peptides that are unmodified or methylated at K4 (ref. 85). The first three N-terminal residues of H3 are involved in the majority of direct contacts, which are also conserved in the complexes of WDR5 with unmodified H3 and H3K4me86–89. The side chain of R2me2s inserts deeply into the open channel of the β-propeller, with the methylated guanidine moiety being sandwiched between two phenylalanine rings85 (Fig. 2d). Notably, in the complexes of WDR5 with various H3 peptides, R2 is bound in the same pocket and adopts a similar conformation; however, Rme2s makes tighter hydrophobic interactions, whereas unmethylated R2 forms an additional water-mediated hydrogen bond85–88.
Functional significance of Rme recognition
Very little is known about the significance of methylarginine readout in histone proteins. One of the few reported examples involves recognition of H3R17me2a and H4R3me2a by the TDRD3 Tudor domain, which facilitates recruitment of this co-transcriptional activator to gene promoters and is necessary for proper coactivation90. The ADD domain of the DNMT3a DNA methyltransferase has been shown to interact with H4R3me2s, promoting silencing of the human β-globin locus91. The association of the WDR5 WD40 module with H3R2me2s—which is produced by PRMT5 and PRMT7—is important for euchromatin maintenance85. Together, these examples demonstrate that recognition of methylated arginines in histone proteins can influence transcription processes.
Acetylation readers
Besides being methylated, lysine can also be acetylated at the ε-amino group. Unlike methylation, this modification changes the electrostatic properties of histone proteins by neutralizing the charge of lysine. At present, lysine acetylation has been found to occur on H3 (K4, K9, K14, K18, K23, K27, K36 and K56), H4 (K5, K8, K12, K16, K20 and K91), H2A (K5 and K9) and H2B (K5, K12, K15, K16, K20 and K120). In general, this PTM leads to a more open chromatin structure, as it weakens the interaction with the negatively charged DNA and is largely associated with a transcriptionally active state. To date, three histone effectors capable of reading acetylated lysine have been identified.
The bromodomain is the most thoroughly characterized acetyllysine reader15,92. Despite little sequence similarity between family members, bromodomains fold into a highly conserved four-helix bundle structure (helices αA, αB, αC and αZ). The inter-helical ZA and BC loops create a deep, primarily hydrophobic cavity into which the acetyllysine inserts. Acetylated lysine makes several contacts with hydrophobic residues, including two often conserved tyrosines, and is stabilized by a hydrogen bond with a highly conserved asparagine. The surface properties of bromodomains vary considerably, accounting for a wide array of ligands93. In general, isolated bromodomains bind to acetylated histones weakly, yet at times they display specificity, imparted by interaction with surrounding residues. Simultaneous recognition of multiply acetylated sequences, either by a single bromodomain or by covalently linked bromodomains, can substantially enhance binding, as seen in TAF1 and Brdt94,95. Two bromodomains in the double bromodomain (DBD) of TAF1 create a V-shaped structure, with both acetyllysine binding pockets oriented in the same direction and separated by ~25 Å, a distance ideal for recognition of a doubly acetylated H4 tail94. Consistently, the TAF1 DBD shows a much higher affinity for H4K5acK12ac and H4K8acK16ac peptides than for monoacetylated peptides, suggesting that each bromodomain interacts with one acetyllysine in the same peptide. Brdt utilizes a different mechanism to increase binding. The first bromodomain of Brdt accommodates both acetyllysines of H4K5acK8ac in a wider hydrophobic pocket (Fig. 3a), whereas the second bromodomain binds H3K18ac95.
Figure 3.

Molecular mechanisms for the recognition of acetyllysine, phosphoserine or phosphothreonine and unmodified histone H3. (a–c) Structures of the readers bound to histone peptides that are acetylated at lysine residues (a), phosphorylated at serine or threonine residues (b) and unmodified (c). Binding-site residues are colored green (a), pink (b) and wheat (c) with the corresponding peptides in orange, blue and purple, respectively. PDB codes: 1E6I, 2KWJ, 2WP2, 4A0N, 2AZM, 2C1N, 3L41, 3A1B, 2YBA and 2PUY.
More recently, the double PHD finger (DPF) of Dpf3b has been found to associate with acetylated histone peptides96,97. The PHD modules in DPF adopt a typical zinc-finger topology but have a large interface, which results in an overall unique scaffold97. The structure of the DPF–H3K14ac complex reveals that the PHD modules are engaged in coordinated interaction97. Whereas the first four N-terminal residues of H3K14ac are bound by the second PHD module, the first PHD module anchors K14ac in the binding pocket composed of hydrophobic and charged residues, including the necessary aspartate-phenylalanine sequence.
The double pleckstrin homology (PH) domain of the histone chaperone Rtt106 has recently been implicated in binding to H3K56ac98. Each PH domain consists of a seven-stranded β-barrel, capped by one or two α-helixes at one of the open ends. The opposite end of the barrel is framed by three variable loops, which, in a canonical PH domain, comprise the binding site for a phosphoinositide lipid99. Although it is unclear whether Rtt106 binds lipids, chemical-shift perturbation analysis reveals that the H3K56ac-binding pocket is located in the second PH module at the interface of the C-terminal α-helix and the β-barrel, with an aspartate, tyrosine and arginine likely forming a cleft for acetylated lysine98.
Functional significance of Kac recognition
For a long time, acetylation of histones was considered to be merely a mechanism for chromatin opening by way of disrupting the association between histones and DNA; however, a number of effector-harboring proteins have been found to bind acetylated lysine. Many of these interactions are implicated in gene transcription, linking the direct readout of this PTM to transcriptional control. The BRD family of proteins is composed of transcription regulators that possess bromodomains, which are specific for singly or multiply acetylated histone peptides93. Activation and regulation of the DPF3b and MOZ target genes requires binding of their DPFs to H3K14ac97,100.
Recognition of H3K56ac by the double PH domain of Rtt106 is essential in gene silencing as well as DNA damage response98. Likewise, association of the bromodomain of BRG1 (the catalytic sub-unit of SWI/SNF) with acetylated H3 is necessary for DNA repair101. Yeast DBD-containing proteins Bdf1 and Bdf2 target H4Kac and regulate transcription and chromatin dynamics, but are also important for replication-coupled DNA repair and normal mRNA splicing102–105. The bromodomain-harboring protein ACF1 is enriched in replicating pericentromeric heterochromatin, and its depletion causes a delay in cell-cycle progression106. A closely related protein, BPTF, contains a bromodomain that binds H4K16ac, which is important for the Hox gene localization of the BPTF–NURF complex107.
Phosphorylation readers
Similar to lysine acetylation, phosphorylation of serine and threonine alters the electrostatic and topographic properties of histones by adding a bulky, negatively charged group to the modified residue. Recognition of phosphorylated sequences by cytosolic proteins has been characterized relatively well; however, much less is known about histone phosphorylation. Histones can be phosphorylated at T3, T6, S10, T11, S28 and T45 of H3; S1 of H4; S1 and T120 of H2A; S139 of human histone variant H2AX and S14 of H2B. These PTMs are essential in DNA damage response pathways, mitosis and transcriptional regulation.
The tandem BRCT domain has been found to specifically recognize phosphorylated S139 of H2AX (H2AXS139ph, also referred to as γH2AX)108. Each BRCT has an αβ sandwich architecture, with a central four-stranded β-sheet surrounded by α-helices on either side (Fig. 3b). The tandem repeats align in a head-to-tail manner, forming a considerable hydrophobic interface. The H2AXS139ph peptide lies in the groove at the interface of two repeats. The phosphorylated S139 residue is held in a basic site through direct and water-mediated hydrogen bonds and ionic interactions. In all characterized BRCT domains, specificity is defined by a hydrophobic residue at the +3 position, which is bound in a well-formed hydrophobic pocket on the protein surface108,109. Additionally, the carboxyl group at the C terminus of γH2AX (at the +3 position) is stabilized by hydrogen bonds with a conserved arginine, contributing substantially to the strength and specificity of the interaction.
The 14-3-3ζ isoform recognizes H3S10ph and H3S28ph110. 14-3-3ζ belongs to a family of 14-3-3 proteins that generally interact with phosphorylated nonhistone targets. The crystal structure of 14-3-3ζ bound to H3S10ph provides insight into the mechanism underlying specificity for the histone ligand. The H3S10ph peptide assumes a partially extended conformation and inserts into a cleft formed by five helices of the U-shaped, nine-helical protein. S10ph is restrained through multiple hydrogen bonding and ionic contacts with two arginine residues and a tyrosine, whereas the G12–G13 sequence adopts a conformation necessary for the C terminus of the peptide to exit the cleft.
The BIR domain of the chromosomal passenger complex subunit Survivin is the newest histone reader that binds H3T3ph111–113. BIR folds into a small αβ structure stabilized by the coordination of a single zinc ion. The H3T3ph peptide is bound in an extended conformation across a largely electronegative surface of the BIR domain, with T3ph placed in a small, positively charged patch composed of a lysine and a histidine. The peptide is anchored through an extensive network of hydrogen bonds involving several aspartate and glutamate residues of BIR and the free amino group of A1, the side chain of K4 and the backbone of the peptide.
Functional significance of Sph and Tph recognition
One of the best-characterized histone phosphorylation marks is on S139 of mammalian and S129 of yeast histone H2A(X). In response to DNA damage, various kinases involved in the DNA repair pathway (ATM, ATR and DNA-PK) phosphorylate this residue to facilitate the repair process. Apart from the tandem BRCT domains of MDC1, 53BP1 and Crb2 (refs. 108,114,115), other proteins implicated in the DNA repair pathway have also been suggested to bind this PTM 108,116,117. Likewise, a proapoptotic function has been proposed for the recognition of adjacent phosphorylated Y142 of H2AX by the PTB domain of Fe65 (ref. 118).
Two other established functions for histone serine or threonine phosphorylation are regulation of mitosis and transcription. The Aurora B kinase phosphorylates H3S10 during mitosis, and this phosphorylation is necessary for chromosome condensation and segregation119. This modification acts to release HP1 proteins from chromatin by disrupting the interaction of their chromodomains with H3K9me3 (refs. 119,120). Survivin binds H3T3ph through its BIR domain to allow proper Aurora B activity at inner centromeres111,121. The 14-3-3 family of proteins has many isoforms, some of which recognize H3S10ph and H3S28ph110. Moreover, this binding is reinforced by acetylation of K14 on the same histone tail122,123. The recruitment of 14-3-3 or its yeast homologs Bmh1 and Bmh2 is necessary for transcription activation of GAL1 and HDAC1, among other genes122,123.
Unmodified H3 readers
A large number of PHD fingers, as well as the ADD and WD40 modules, associate with the unmodified H3 tail (Fig. 3c). Some similarity in the molecular mechanisms for the recognition of unmodified H3 and H3K4me3 by the PHD fingers is apparent. Like H3K4me3, the unmodified H3 peptide forms the third antiparallel β-strand in the complex124. The side chain of A1 is buried in a small hydrophobic cavity, and the N-amino group of A1 is fixed by hydrogen bonds with backbone carbonyls of the protein. The guanidinium moiety of R2 is commonly restrained through the formation of hydrogen bonding and ionic contacts. Differences between the two binding mechanisms arise from the distinct coordination of K4. The unmodified H3–specific PHD fingers lack the aromatic cage, and instead, unmethylated K4 is bound by a set of hydrogen bonds and salt bridges formed with the acidic residues clustered on the surface of the protein. The majority of PHD fingers interact with a long stretch of the unmodified H3 tail, recognizing up to nine residues of H3 (ref. 44); however, the extended PHD finger of UHRF1 makes contacts with only the first four residues of unmodified H3 (refs. 125–127). The R2 residue of the peptide, rather than K4, is robustly bound by several hydrogen bonds, which results in a decreased sensitivity to PTMs on K4.
The ADD domain consists of a GATA-like C4 finger followed by a PHD finger, which together coordinate three zinc ions. The ADD domain binds to the unmodified H3 peptide, using a mechanism typical of PHD fingers and recognizing seven residues of the peptide128. The WD40 domain of Nurf55 is in direct contact with nine residues of unmodified H3 (ref. 67). The structure of the Nurf55–unmodified H3 complex reveals a hydrophobic cavity accommodating A1 and several hydrogen bonds fixing the amino groups of A1 and K4. However, buried within the propeller fold, R2 is not stabilized through hydrogen bonds and is instead sandwiched between two aromatic residues.
Combinatorial readout of and cross-talk between PTMs
The list of newly identified histone readers has grown rapidly, yet the finite number of readers and the numerous biological processes they mediate suggest that explicit mechanisms exist to differentiate functions of the effector-containing proteins and elicit distinct biological outcomes. Given the extensive and complex nature of the chromatin landscape, several mechanisms involving the combinatorial readout of epigenetic marks have been uncovered.
The activity of a reader toward a particular modification can be influenced by neighboring PTMs (Fig. 4a). In fact, many readers associate with a substantial stretch of the histone tail, allowing for the sensing of multiple marks. The neighboring PTMs can enhance or impede interaction with the target PTM. The classic example is the inhibition of binding of HP1 chromodomain to H3K9me3 by phosphorylation of S10, which may negatively regulate localization of HP1 at chromatin during mitosis120. Similarly, phosphorylation of T3 decreases affinity of CHD1 DCD for H3K4me3 (ref. 46). Methylation of H3K4 abrogates interactions of the unmodified H3–specific PHD fingers44 and ADD51, whereas acetylation of H4K8 increases binding of the Brdt bromodomain to H4K5ac95. Methylation of R2 enhances interaction of the Rag2 PHD finger with H3K4me3 (ref. 129) but inhibits the histone binding activities of TAF3 and AIRE PHD fingers59,130.
Figure 4.
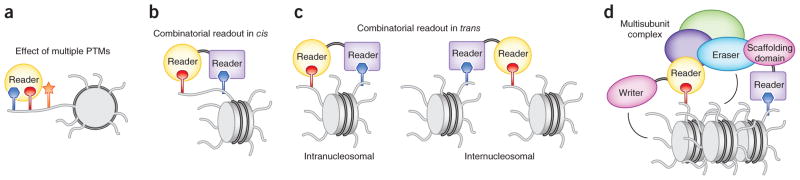
Combinatorial readout of PTMs. (a–c) Recognition of a target PTM is influenced by adjacent PTMs on the same histone tail (a) and the combined action of multiple readers within the same protein (b,c). (d) Multivalent engagement of readers within individual subunits of the complex. The reader-harboring proteins can also contain the catalytic domains (which act as writers and erasers) or scaffolding domains that bridge their host proteins with other subunits of the complex. Readers can recognize PTMs on a single histone tail (cis mechanism) or different histone tails (trans mechanism).
Recruitment of a reader to a specific genomic region can be further modulated through the combinatorial action of multiple effectors (Fig. 4b,c). A number of chromatin-associated proteins contain more than one reader, in the form of several copies of the same reader or a combination of various readers that are often specific for distinct PTMs. The combinatorial readout in cis, where effectors bind PTMs within the same histone tail (Fig. 4b), was initially reported for the TAF1 DBD94. Each bromodomain of the TAF1 DBD recognizes one acetyllysine on the same diacetylated H4 peptide. Coincidence detection of two histone PTMs by different effectors has been demonstrated for the PHD-bromodomain cassettes of TRIM24 and TRIM33 (refs. 55,131). The PHD finger of TRIM33 binds to K9me3, and the bromodomain recognizes K18ac on the same H3 peptide55. Likewise, TRIM24 concomitantly associates with unmodified H3 and K23ac of the H3 tail through its PHD finger and bromodomain, respectively131. The PHD finger and TTD of UHRF1, a key factor for maintenance of DNA methylation, synergistically recognize unmodified H3 and K9me3 of the H3K9me3 peptide132. Combinatorial reading of PTMs by an effector and a catalytic histone-binding domain has been shown for the histone demethylases PHF8 and KIAA1718, where binding of the PHD finger to H3K4me3 in the doubly methylated H3K4me3K9me2 and H3K4me3K27me2 peptides directs association of the catalytic domains with K9me2 and K27me2, respectively60. BRPF1/2, a member of the trithorax family with a central role during development, contains two PHD fingers, a PWWP domain and a bromodomain. Although it has not yet been determined whether the interactions occur on the same histone tail, the PWWP domain preferentially binds H3K36me3 and the first PHD domain associates with unmodified H3 (refs. 35,36,133,134).
Multiple PTMs can also be recognized on separate tails of a single nucleosome or on adjacent nucleosomes that are directly linked by DNA or are otherwise in close spatial proximity (Fig. 4c). Despite the widespread phenomenon of multivalent engagement, characterization of combinatorial readout in trans poses unique challenges, and only a few examples have thus far been described. BPTF, a subunit of the NURF chromatin-remodeling complex, contains an H3K4me3-specific PHD finger and an acetyllysine-binding bromodomain, connected by a rigid α-helical linker40,43. A modeling study has revealed that the PHD-linker-bromodomain assembly fits complementarily on the surface of a single nucleosome with the PHD finger and bromo-domain concomitantly interacting with H3K4me3 and H4K16ac, respectively135. More recently, the intranucleosomal engagement of the two readers and coexistence of the H3K4me3 and H4K16ac marks on a single nucleosome within human cells have been confirmed experimentally107. Another example of the in _trans_–acting readers is the PHD fingers of CHD4, the interdomain organization of which positions these two modules with a duplicate function to concurrently associate with two H3 tails of a nucleosome136.
An even more complex combinatorial trans readout can be seen in chromatin complexes containing histone readers in multiple subunits (Fig. 4d). The interplay between such effectors generates a multifaceted network of intertwined contacts that can provide a high degree of specificity. The PHD finger–containing subunits of the HBO1 complex work together to regulate its acetyltransferase activity137,138. Similarly, the methyltransferase function of the PRC2 complex is modulated by its subunits harboring WD40 repeats, Tudor and PHD fingers139. The SAGA complex, which has a major role in transcription regulation, contains bromodomains, chromodomains and TTDs that bridge the complex to acetyllysine- and H3K4me3-enriched chromatin140,141. The MYST HATs are also present in complexes harboring multiple histone readers142, including the PHD fingers of BRPF1, ING5 and MOZ138. The tumor suppressor Tip60 HAT complex contains subunits with the H3K4me3-specific PHD domain, H3K36me3- and H3K4me1-binding chromobarrel domains and two bromodomains linked to diacetylated H4 (refs. 41,61,95,143). Furthermore, many nuclear complexes are not static and undergo component swapping, a powerful mechanism for altering their chromatin-targeting capabilities. Beyond mediating specific chromatin anchoring, PTM cross-talk can also trigger a cascading series of writing, erasing and reading events. For instance, the PIM1 kinase phosphorylates H3K9ac, generating H3K9acS10ph144. This mark is recognized by 14-3-3, which recruits the acetyltransferase MOF. MOF, in turn, deposits H4K16ac, a mark targeted by the bromodomain of BRD4, which subsequently recruits P-TEFb, necessary for transcription elongation144.
Concluding remarks
Recognition of PTMs by histone readers has become an important paradigm in chromatin biology. Over the past decade, a wealth of information has been gathered on histone PTMs and PTM-reader relationships, shedding light on the incredibly intricate nature of the chromatin landscape and resultant interactions. The biological consequences of effector-PTM interactions are highly context dependent, relying on the combinatorial readout of the spatially and temporally fluctuating local epigenetic environment and leading to a highly fine-tuned targeting of particular genomic sites.
Recent significant advances in high-throughput technologies allow for the exploration of interactions between readers and peptide fragments of modified histones at a comprehensive level (Box 1); however, little is known about the activities of these domains in the context of the nucleosome or chromatin fiber. Moreover, the multifaceted contacts between PTMs and readers, the synergistic or antagonistic nature of their interactions and the engagement of other components of the epigenetic machinery are only beginning to be characterized. Future studies will provide significant insight into the epigenetic regulatory mechanisms and may lead to the discoveries of new pharmacological targets and biomarkers. For example, recently developed small-molecule inhibitors for the bromodomains of the BET protein family show anticancer activity and demonstrate promising results in preclinical studies145. Unlike mutations in the genome, epimutations are largely reversible, and thus epigenetic therapy has a high therapeutic potential. Although in-depth characterization of the epigenetic pathways is necessary to better understand and exploit this potential, the prospective benefits are far reaching.
BOX 1. Methodologies for the identification of histone readers.
Novel techniques are being developed for rapid high-throughput and discovery-based screening of novel PTM readers and characterization of the effect of adjacent PTMs.
Histone microarrays146–149
Histone peptides carrying various PTMs are synthesized on cellulose support (SPOT and CelluSpot) or attached to streptavidin-coated platforms by a biotin tag. GST- or His-tagged protein is incubated with the screen, then the bound fraction is detected by western blot or fluorescence. Binding to a single PTM or combinations of PTMs can be assayed in a high-throughput manner; however, this technique requires purification of the protein of interest, and initial generation of the screen can be time consuming.
Bead-based approach150
This approach utilizes a PTM-randomized combinatorial library of histone peptides. Proteins incubated with the library are captured on beads and their target peptides are analyzed by MS. This method is very powerful in detecting synergistic and inhibitory combinations of known and yet-to-be-discovered PTMs, but it has the same limitations as histone microarrays.
Stable isotope labeling by amino acids in cell culture (SILAC) technolog151,152
Nuclear extracts from cells grown in SILAC medium are incubated with PTM-bearing immobilized histone peptides or recombinant nucleosomes. Bound proteins or protein complexes are pulled down and analyzed by quantitative MS. This approach provides an unbiased way for the identification of new PTM readers; however, the particular protein and domain responsible for the targeting may not be easily identified.
Acknowledgments
Research in T.G.K.’s laboratory is supported by grants from the US National Institutes of Health (NIH; GM096863 and CA113472 to T.G.K.). Work in J.C.’s laboratory is supported by grants from the Canadian Institutes of Health Research (MOP-14308 and MOP-64289 to J.C.). M.-E.L. is supported by a Fonds de recherche du Québec–Santé studentship and J.C. is a Canada Research Chair. C.A.M. is supported by an NIH National Research Service Award postdoctoral fellowship (F32 HL096399) and the Cancer League of Colorado.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8-Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 4.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 5.Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–637. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 7.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 9.Brownell JE, et al. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 10.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 11.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 12.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 13.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–616. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhalluin C, et al. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 16.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bannister AJ, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 18.Nielsen PR, et al. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature. 2002;416:103–107. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- 19.Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9–methylated histone H3 tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 20.Fischle W, et al. Molecular basis for the discrimination of repressive methyllysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003;17:1823–1828. doi: 10.1101/gad.269603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blus BJ, Wiggins K, Khorasanizadeh S. Epigenetic virtues of chromodomains. Crit Rev Biochem Mol Biol. 2011;46:507–526. doi: 10.3109/10409238.2011.619164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ballaré C, et al. Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nat Struct Mol Biol. 2012 Oct 28; doi: 10.1038/ nsmb.2434. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Botuyan MV, et al. Structural basis for the methylation state–specific recognition of histone H4–K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine 4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 26.Lee J, Thompson JR, Botuyan MV, Mer G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat Struct Mol Biol. 2008;15:109–111. doi: 10.1038/nsmb1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Musselman CA, et al. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat Struct Mol Biol. 2012;19:aaa–bbb. doi: 10.1038/nsmb.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bian C, et al. Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation. EMBO J. 2011;30:2829–2842. doi: 10.1038/emboj.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Min J, et al. L3MBTL1 recognition of mono- and dimethylated histones. Nat Struct Mol Biol. 2007;14:1229–1230. doi: 10.1038/nsmb1340. [DOI] [PubMed] [Google Scholar]
- 30.Li H, et al. Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Mol Cell. 2007;28:677–691. doi: 10.1016/j.molcel.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grimm C, et al. Structural and functional analyses of methyllysine binding by the malignant brain tumour repeat protein Sex comb on midleg. EMBO Rep. 2007;8:1031–1037. doi: 10.1038/sj.embor.7401085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo Y, et al. Methylation state–specific recognition of histones by the MBT repeat protein L3MBTL2. Nucleic Acids Res. 2009;37:2204–2210. doi: 10.1093/nar/gkp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klymenko T, et al. A Polycomb group protein complex with sequence-specific DNA-binding and selective methyllysine–binding activities. Genes Dev. 2006;20:1110–1122. doi: 10.1101/gad.377406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, et al. Regulation of Set9-mediated H4K20 methylation by a PWWP domain protein. Mol Cell. 2009;33:428–437. doi: 10.1016/j.molcel.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vezzoli A, et al. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat Struct Mol Biol. 2010;17:617–619. doi: 10.1038/nsmb.1797. [DOI] [PubMed] [Google Scholar]
- 36.Wu H, et al. Structural and histone binding ability characterizations of human PWWP domains. PLoS ONE. 2011;6:e18919. doi: 10.1371/journal.pone.0018919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang P, et al. Structure of human MRG15 chromodomain and its binding to Lys36-methylated histone H3. Nucleic Acids Res. 2006;34:6621–6628. doi: 10.1093/nar/gkl989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu C, Cui G, Botuyan MV, Mer G. Structural basis for the recognition of methylated histone H3K36 by the Eaf3 subunit of histone deacetylase complex Rpd3S. Structure. 2008;16:1740–1750. doi: 10.1016/j.str.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim D, et al. Corecognition of DNA and a methylated histone tail by the MSL3 chromodomain. Nat Struct Mol Biol. 2010;17:1027–1029. doi: 10.1038/nsmb.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, et al. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peña PV, et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi X, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wysocka J, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 44.Musselman CA, Kutateladze TG. Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 2011;39:9061–9071. doi: 10.1093/nar/gkr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He F, et al. Structural insight into the zinc-finger CW domain as a histone modification reader. Structure. 2010;18:1127–1139. doi: 10.1016/j.str.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 46.Flanagan JF, et al. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 47.Margueron R, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu C, et al. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2) Proc Natl Acad Sci USA. 2010;107:19266–19271. doi: 10.1073/pnas.1008937107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Collins RE, et al. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat Struct Mol Biol. 2008;15:245–250. doi: 10.1038/nsmb.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuo AJ, et al. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature. 2012;484:115–119. doi: 10.1038/nature10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iwase S, et al. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat Struct Mol Biol. 2011;18:769–776. doi: 10.1038/nsmb.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eustermann S, et al. Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat Struct Mol Biol. 2011;18:777–782. doi: 10.1038/nsmb.2070. [DOI] [PubMed] [Google Scholar]
- 53.Musselman CA, et al. Binding of the CHD4 PHD2 finger to histone H3 is modulated by covalent modifications. Biochem J. 2009;423:179–187. doi: 10.1042/BJ20090870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mansfield RE, et al. Plant homeodomain (PHD) fingers of CHD4 are histone H3-binding modules with preference for unmodified H3K4 and methylated H3K9. J Biol Chem. 2011;286:11779–11791. doi: 10.1074/jbc.M110.208207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xi Q, et al. A poised chromatin platform for TGF-β access to master regulators. Cell. 2011;147:1511–1524. doi: 10.1016/j.cell.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schneider R, et al. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- 57.Bernstein BE, et al. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci USA. 2002;99:8695–8700. doi: 10.1073/pnas.082249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang G, et al. Distinct localization of histone H3 acetylation and H3–K4 methylation to the transcription start sites in the human genome. Proc Natl Acad Sci USA. 2004;101:7357–7362. doi: 10.1073/pnas.0401866101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vermeulen M, et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 60.Horton JR, et al. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat Struct Mol Biol. 2010;17:38–43. doi: 10.1038/nsmb.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jeong KW, et al. Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat Struct Mol Biol. 2011;18:1358–1365. doi: 10.1038/nsmb.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bannister AJ, et al. Spatial distribution of di- and trimethyl lysine 36 of histone H3 at active genes. J Biol Chem. 2005;280:17732–17736. doi: 10.1074/jbc.M500796200. [DOI] [PubMed] [Google Scholar]
- 63.Carrozza MJ, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 64.Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3–K36 links histone deacetylation to Pol II elongation. Mol Cell. 2005;20:971–978. doi: 10.1016/j.molcel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 65.Keogh MC, et al. Cotranscriptional Set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 66.Venkatesh S, et al. Set2-mediated histone H3 lysine36 methylation suppresses histone exchange on transcribed genes. Nature. 2012 doi: 10.1038/nature11326. (in the press) [DOI] [PubMed] [Google Scholar]
- 67.Schmitges FW, et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell. 2011;42:330–341. doi: 10.1016/j.molcel.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 68.Yuan W, et al. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem. 2011;286:7983–7989. doi: 10.1074/jbc.M110.194027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luco RF, et al. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alekseyenko AA, et al. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell. 2008;134:599–609. doi: 10.1016/j.cell.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maltby VE, et al. Histone H3 lysine 36 methylation targets the Isw1b remodeling complex to chromatin. Mol Cell Biol. 2012;32:3479–3485. doi: 10.1128/MCB.00389-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smolle M, et al. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat Struct Mol Biol. 2012;19:884–892. doi: 10.1038/nsmb.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Daugaard M, et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat Struct Mol Biol. 2012;19:803–810. doi: 10.1038/nsmb.2314. [DOI] [PubMed] [Google Scholar]
- 74.Biswas D, et al. A role for Chd1 and Set2 in negatively regulating DNA replication in Saccharomyces cerevisiae. Genetics. 2008;178:649–659. doi: 10.1534/genetics.107.084202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pryde F, et al. H3 k36 methylation helps determine the timing of cdc45 association with replication origins. PLoS ONE. 2009;4:e5882. doi: 10.1371/journal.pone.0005882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mallette FA, et al. RNF8- and RNF168-dependent degradation of KDM4A/ JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012;31:1865–1878. doi: 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peters AH, et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell. 2003;12:1577–1589. doi: 10.1016/s1097-2765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- 78.Rice JC, et al. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell. 2003;12:1591–1598. doi: 10.1016/s1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- 79.Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 80.Canzio D, et al. Chromodomain-mediated oligomerization of HP1 suggests a nucleosome-bridging mechanism for heterochromatin assembly. Mol Cell. 2011;41:67–81. doi: 10.1016/j.molcel.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen C, Nott TJ, Jin J, Pawson T. Deciphering arginine methylation: Tudor tells the tale. Nat Rev Mol Cell Biol. 2011;12:629–642. doi: 10.1038/nrm3185. [DOI] [PubMed] [Google Scholar]
- 82.Yang Y, et al. TDRD3 is an effector molecule for arginine-methylated histone marks. Mol Cell. 2010;40:1016–1023. doi: 10.1016/j.molcel.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu K, et al. Crystal structure of TDRD3 and methyl-arginine binding characterization of TDRD3, SMN and SPF30. PLoS ONE. 2012;7:e30375. doi: 10.1371/journal.pone.0030375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tripsianes K, et al. Structural basis for dimethylarginine recognition by the Tudor domains of human SMN and SPF30 proteins. Nat Struct Mol Biol. 2011;18:1414–1420. doi: 10.1038/nsmb.2185. [DOI] [PubMed] [Google Scholar]
- 85.Migliori V, et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat Struct Mol Biol. 2012;19:136–144. doi: 10.1038/nsmb.2209. [DOI] [PubMed] [Google Scholar]
- 86.Couture JF, Collazo E, Trievel RC. Molecular recognition of histone H3 by the WD40 protein WDR5. Nat Struct Mol Biol. 2006;13:698–703. doi: 10.1038/nsmb1116. [DOI] [PubMed] [Google Scholar]
- 87.Ruthenburg AJ, et al. Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat Struct Mol Biol. 2006;13:704–712. doi: 10.1038/nsmb1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schuetz A, et al. Structural basis for molecular recognition and presentation of histone H3 by WDR5. EMBO J. 2006;25:4245–4252. doi: 10.1038/sj.emboj.7601316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Han Z, et al. Structural basis for the specific recognition of methylated histone H3 lysine 4 by the WD-40 protein WDR5. Mol Cell. 2006;22:137–144. doi: 10.1016/j.molcel.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 90.Yang Y, et al. TDRD3 is an effector molecule for arginine-methylated histone marks. Mol Cell. 2010;40:1016–1023. doi: 10.1016/j.molcel.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao Q, et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol. 2009;16:304–311. doi: 10.1038/nsmb.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sanchez R, Zhou MM. The role of human bromodomains in chromatin biology and gene transcription. Curr Opin Drug Discov Devel. 2009;12:659–665. [PMC free article] [PubMed] [Google Scholar]
- 93.Filippakopoulos P, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jacobson RH, Ladurner AG, King DS, Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science. 2000;288:1422–1425. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- 95.Morinière J, et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature. 2009;461:664–668. doi: 10.1038/nature08397. [DOI] [PubMed] [Google Scholar]
- 96.Lange M, et al. Regulation of muscle development by DPF3, a novel histone acetylation and methylation reader of the BAF chromatin remodeling complex. Genes Dev. 2008;22:2370–2384. doi: 10.1101/gad.471408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zeng L, et al. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature. 2010;466:258–262. doi: 10.1038/nature09139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Su D, et al. Structural basis for recognition of H3K56-acetylated histone H3–H4 by the chaperone Rtt106. Nature. 2012;483:104–107. doi: 10.1038/nature10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kutateladze TG. Translation of the phosphoinositide code by PI effectors. Nat Chem Biol. 2010;6:507–513. doi: 10.1038/nchembio.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Qiu Y, et al. Combinatorial readout of unmodified H3R2 and acetylated H3K14 by the tandem PHD finger of MOZ reveals a regulatory mechanism for HOXA9 transcription. Genes Dev. 2012;26:1376–1391. doi: 10.1101/gad.188359.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee HS, Park JH, Kim SJ, Kwon SJ, Kwon J. A cooperative activation loop among SWI/SNF, γ-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J. 2010;29:1434–1445. doi: 10.1038/emboj.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Albulescu LO, et al. A quantitative, high-throughput reverse genetic screen reveals novel connections between pre-mRNA splicing and 5′- and 3′-end transcript determinants. PLoS Genet. 2012;8:e1002530. doi: 10.1371/journal.pgen.1002530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Koerber RT, Rhee HS, Jiang C, Pugh BF. Interaction of transcriptional regulators with specific nucleosomes across the Saccharomyces genome. Mol Cell. 2009;35:889–902. doi: 10.1016/j.molcel.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Garabedian MV, et al. The double-bromodomain proteins Bdf1 and Bdf2 modulate chromatin structure to regulate S-phase stress response in Schizosaccharomyces pombe. Genetics. 2012;190:487–500. doi: 10.1534/genetics.111.135459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Matangkasombut O, Buratowski S. Different sensitivities of bromodomain factors 1 and 2 to histone H4 acetylation. Mol Cell. 2003;11:353–363. doi: 10.1016/s1097-2765(03)00033-9. [DOI] [PubMed] [Google Scholar]
- 106.Collins N, et al. An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat Genet. 2002;32:627–632. doi: 10.1038/ng1046. [DOI] [PubMed] [Google Scholar]
- 107.Ruthenburg AJ, et al. Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell. 2011;145:692–706. doi: 10.1016/j.cell.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stucki M, et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 109.Williams JS, et al. γH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J. 2010;29:1136–1148. doi: 10.1038/emboj.2009.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Macdonald N, et al. Molecular basis for the recognition of phosphorylated and phosphoacetylated histone H3 by 14–3–3. Mol Cell. 2005;20:199–211. doi: 10.1016/j.molcel.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 111.Jeyaprakash AA, Basquin C, Jayachandran U, Conti E. Structural basis for the recognition of phosphorylated histone H3 by the survivin subunit of the chromosomal passenger complex. Structure. 2011;19:1625–1634. doi: 10.1016/j.str.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 112.Du J, Kelly AE, Funabiki H, Patel DJ. Structural basis for recognition of H3T3ph and Smac/DIABLO N-terminal peptides by human Survivin. Structure. 2012;20:185–195. doi: 10.1016/j.str.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Niedzialkowska E, et al. Molecular basis for phosphospecific recognition of histone H3 tails by Survivin paralogs at inner centromeres. Mol Biol Cell. 2012;23:1457–1466. doi: 10.1091/mbc.E11-11-0904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ward IM, Minn K, Jorda KG, Chen J. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J Biol Chem. 2003;278:19579–19582. doi: 10.1074/jbc.C300117200. [DOI] [PubMed] [Google Scholar]
- 115.Sofueva S, Du LL, Limbo O, Williams JS, Russell P. BRCT domain interactions with phospho-histone H2A target Crb2 to chromatin at double-strand breaks and maintain the DNA damage checkpoint. Mol Cell Biol. 2010;30:4732–4743. doi: 10.1128/MCB.00413-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Stucki M, Jackson SP. γH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst ) 2006;5:534–543. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 117.Li X, et al. Structure of C-terminal tandem BRCT repeats of Rtt107 protein reveals critical role in interaction with phosphorylated histone H2A during DNA damage repair. J Biol Chem. 2012;287:9137–9146. doi: 10.1074/jbc.M111.311860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cook PJ, et al. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458:591–596. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hirota T, Lipp JJ, Toh BH, Peters JM. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438:1176–1180. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- 120.Fischle W, et al. Regulation of HP1–chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–1122. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- 121.Wang F, et al. Histone H3 Thr-3 phosphorylation by Haspin positions Aurora B at centromeres in mitosis. Science. 2010;330:231–235. doi: 10.1126/science.1189435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Walter W, et al. 14-3-3 interaction with histone H3 involves a dual modification pattern of phosphoacetylation. Mol Cell Biol. 2008;28:2840–2849. doi: 10.1128/MCB.01457-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Winter S, et al. 14-3-3 proteins recognize a histone code at histone H3 and are required for transcriptional activation. EMBO J. 2008;27:88–99. doi: 10.1038/sj.emboj.7601954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lan F, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rajakumara E, et al. PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol Cell. 2011;43:275–284. doi: 10.1016/j.molcel.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang C, et al. Structural basis for site-specific reading of unmodified R2 of histone H3 tail by UHRF1 PHD finger. Cell Res. 2011;21:1379–1382. doi: 10.1038/cr.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hu L, Li Z, Wang P, Lin Y, Xu Y. Crystal structure of PHD domain of UHRF1 and insights into recognition of unmodified histone H3 arginine residue 2. Cell Res. 2011;21:1374–1378. doi: 10.1038/cr.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ooi SK, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ramón-Maiques S, et al. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc Natl Acad Sci USA. 2007;104:18993–18998. doi: 10.1073/pnas.0709170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chignola F, et al. The solution structure of the first PHD finger of autoimmune regulator in complex with non-modified histone H3 tail reveals the antagonistic role of H3R2 methylation. Nucleic Acids Res. 2009;37:2951–2961. doi: 10.1093/nar/gkp166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tsai WW, et al. TRIM24 links a noncanonical histone signature to breast cancer. Nature. 2010;468:927–932. doi: 10.1038/nature09542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Arita K, et al. Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc Natl Acad Sci USA. 2012;109:12950–12955. doi: 10.1073/pnas.1203701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Laue K, et al. The multidomain protein Brpf1 binds histones and is required for Hox gene expression and segmental identity. Development. 2008;135:1935–1946. doi: 10.1242/dev.017160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Qin S, et al. Recognition of unmodified histone H3 by the first PHD finger of bromodomain-PHD finger protein 2 provides insights into the regulation of histone acetyltransferases monocytic leukemic zinc-finger protein (MOZ) and MOZ-related factor (MORF) J Biol Chem. 2011;286:36944–36955. doi: 10.1074/jbc.M111.244400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Musselman CA, et al. Bivalent recognition of nucleosomes by the tandem PHD fingers of the CHD4 ATPase is required for CHD4-mediated repression. Proc Natl Acad Sci USA. 2012;109:787–792. doi: 10.1073/pnas.1113655109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hung T, et al. ING4 mediates cross-talk between histone H3 K4 trimethylation and H3 acetylation to attenuate cellular transformation. Mol Cell. 2009;33:248–256. doi: 10.1016/j.molcel.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Saksouk N, et al. HBO1 HAT complexes target chromatin throughout gene coding regions via multiple PHD finger interactions with histone H3 tail. Mol Cell. 2009;33:257–265. doi: 10.1016/j.molcel.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hassan AH, et al. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell. 2002;111:369–379. doi: 10.1016/s0092-8674(02)01005-x. [DOI] [PubMed] [Google Scholar]
- 141.Bian C, et al. Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation. EMBO J. 2011;30:2829–2842. doi: 10.1038/emboj.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Avvakumov N, Cote J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene. 2007;26:5395–5407. doi: 10.1038/sj.onc.1210608. [DOI] [PubMed] [Google Scholar]
- 143.Xie L, et al. KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. EMBO J. 2011;30:1473–1484. doi: 10.1038/emboj.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zippo A, et al. Histone cross-talk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell. 2009;138:1122–1136. doi: 10.1016/j.cell.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 145.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nady N, Min J, Kareta MS, Chedin F, Arrowsmith CHA. SPOT on the chromatin landscape? Histone peptide arrays as a tool for epigenetic research. Trends Biochem Sci. 2008;33:305–313. doi: 10.1016/j.tibs.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 147.Fuchs SM, Krajewski K, Baker RW, Miller VL, Strahl BD. Influence of combinatorial histone modifications on antibody and effector protein recognition. Curr Biol. 2011;21:53–58. doi: 10.1016/j.cub.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bua DJ, et al. Epigenome microarray platform for proteome-wide dissection of chromatin-signaling networks. PLoS ONE. 2009;4:e6789. doi: 10.1371/journal.pone.0006789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bock I, et al. Application of Celluspots peptide arrays for the analysis of the binding specificity of epigenetic reading domains to modified histone tails. BMC Biochem. 2011;12:48. doi: 10.1186/1471-2091-12-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Garske AL, et al. Combinatorial profiling of chromatin binding modules reveals multisite discrimination. Nat Chem Biol. 2010;6:283–290. doi: 10.1038/nchembio.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vermeulen M, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 152.Bartke T, et al. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell. 2010;143:470–484. doi: 10.1016/j.cell.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Brien GL, et al. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat Struct Mol Biol. 2012 Nov 18; doi: 10.1038/nsmb.2449. advance online publication. [DOI] [PubMed] [Google Scholar]