RAS Interaction with PI3K: More Than Just Another Effector Pathway (original) (raw)
Abstract
RAS proteins are small GTPases known for their involvement in oncogenesis: around 25% of human tumors present mutations in a member of this family. RAS operates in a complex signaling network with multiple activators and effectors, which allows them to regulate many cellular functions such as cell proliferation, differentiation, apoptosis, and senescence. Phosphatidylinositol 3-kinase (PI3K) is one of the main effector pathways of RAS, regulating cell growth, cell cycle entry, cell survival, cytoskeleton reorganization, and metabolism. However, it is the involvement of this pathway in human tumors that has attracted most attention. PI3K has proven to be necessary for RAS-induced transformation in vitro, and more importantly, mice with mutations in the PI3K catalytic subunit p110α that block its ability to interact with RAS are highly resistant to endogenous oncogenic KRAS-induced lung tumorigenesis and HRAS-induced skin carcinogenesis. These animals also have a delayed development of the lymphatic vasculature. Many PI3K inhibitors have been developed that are now in clinical trials. However, it is a complex pathway with many feedback loops, and interactions with other pathways make the results of its inhibition hard to predict. Combined therapy with another RAS-regulated pathway such as RAF/MEK/ERK may be the most effective way to treat cancer, at least in animal models mimicking the human disease. In this review, we will summarize current knowledge about how RAS regulates one of its best-known effectors, PI3K.
Keywords: Ras, PI3-kinase, lung cancer, lymphangiogenesis
Introduction
The RAS family of GTPases (HRAS, NRAS, and KRAS) comprises proteins that are highly conserved across species and has key roles in numerous basic cellular functions, including control of proliferation, differentiation, and apoptosis. RAS proteins are molecular switches that cycle between 2 conformational states: one when they are bound to GTP, the active form, and another one when bound to GDP, the inactive form.1-4 Guanine nucleotide exchange factors, or GEFs, promote formation of GTP-bound RAS,5 whereas GTPase-activating proteins, or GAPs, stimulate the hydrolysis of GTP on RAS, returning them to their inactive state.6
Study of wild-type RAS proteins has been important to clarify its role in normal developmental processes. However, it is the study of oncogenic RAS signaling that has been essential to establish a molecular basis for the pathogenesis of human cancer,7-10 as it is now clear that activating mutations in members of the RAS family of genes are among the most common genetic lesions in human tumors.11 These mutations lock RAS proteins into a constitutively activated state in which they signal to downstream effectors even in the absence of extracellular stimuli. Involvement of RAS signaling in cancer is accentuated by the incidence not only of RAS mutations but also the deregulation of many of its regulators or effector pathways.12-18
Physiological and oncogenic activation of RAS stimulates a wide range of downstream signaling pathways. The first RAS effector pathway to be identified was the RAF-MEK-ERK pathway.19-22 This pathway is an essential, shared element of mitogenic signaling involving tyrosine kinase receptors, leading to a wide range of cellular responses, including growth, differentiation, inflammation, and apoptosis.23 The RAF family of proteins (Raf-1, A-Raf, and B-Raf) is serine/threonine kinases that bind to the effector region of RAS-GTP, thus inducing translocation of the protein to the plasma membrane. Once there, RAF proteins are activated and phosphorylated by different protein kinases.24-28 Active RAF phosphorylates MEK that, in turn, phosphorylates and activates extracellular signal–regulated kinases 1 and 2 (ERK1/ 2).29 Several studies have supported the importance of this effector pathway by showing the ability of Raf and MEK to transform rodent fibroblasts30 as well as the ability of RAF inhibitors to revert aspects of the RAS-driven transformed phenotype, such as growth in soft agar.31
The second best-characterized RAS effector family is phosphoinositide 3-kinases (PI3Ks), which play important roles as mediators of RAS-mediated cell survival and proliferation.32,33 When active, PI3K converts phosphatidylinositol (4,5)-bisphosphate (PIP2) into phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3, in turn, binds the pleckstrin homology (PH) domain of Akt/PKB, stimulating its kinase activity, resulting in the phosphorylation of a host of other proteins that affect cell growth, cell cycle entry, and cell survival. PI3K can also activate Rac, and this activation is involved in cytoskeleton reorganization.34 The functional importance of the RAS-PI3K pathway will be more extensively discussed in the next sections.
Another well-characterized RAS effector pathway involves Ral-GEF proteins. RalGEF members (RalGDS, RGL, RGL2, and RGL3) link RAS proteins to activation of the RalA and RalB small GTPases.35 The biological function of these proteins is not yet fully understood, although there is evidence that they play an important role of this pathway in RAS-mediated transformation and tumorigenesis in vivo.36,37 Expression of RalGDS cooperates with constitutively activated RAF to induce focus formation, suggesting a cooperation of RalGEF in RAS-mediated transformation in vitro.38
Apart from the above-mentioned effectors, in recent years, an increasing number of molecules that specifically interact with RAS have been described, including Tiam1, p120GAP, NF1, MEKK1, Rin1, AF-6, PKC-ζ, Nore1, Canoe, and others. These proteins bind to RAS exclusively when it is in an activated state39; however, the physiological role of some of these effectors has not yet been found. It has also been described that abnormal RAS signaling causes deregulation of the Rho GTPases family, including Rac1 and RhoA, but no direct association has been found yet.
RAS family members, as well as their effectors, have unique intracellular localizations, and overlapping of these localizations likely restricts many of the interactions. In addition, many of the best-studied RAS effector molecules are members of large families. RAS molecules might be able to regulate different members of the same family, and these selective interactions may have important biological consequences. It should be mentioned that the different RAS isoforms exhibit quantitative and qualitative differences in their ability to activate a particular effector, that some RAS effectors also serve as effectors for other RAS family proteins, and that not all isoforms within a class of effectors have been verified as bona fide RAS effectors.40
PI3K Generalities
It has been more than 20 years since phosphatidylinositol 3-kinase (PI3K) was first discovered. The kinase activity of PI3K was first reported to be associated with viral oncoproteins.41 Subsequent studies employing mouse knockouts of both the regulatory and catalytic subunits of PI3K resulted in a number of deficits including embryonic lethality, B cell defects, liver necrosis, and colorectal cancer.42 The PI3Ks are heterodimeric lipid kinases composed of catalytic and adaptor/regulatory subunit variants encoded by separate genes and alternative splicing. PI3Ks are important regulators of cellular growth, transformation, adhesion, apoptosis, survival, and motility.34,43-45
The PI3K family of enzymes is organized into 3 main classes (class I, II, and III), and various subgroups have been categorized based on their primary structure, substrate specificity, and regulation.32,45 The catalytic subunits for the class I PI3Ks are p110α, p110β, p110γ, and p110δ (which are the products of genes PIK3CA, PIK3CB, PIK3CG, and PIK3CD, respectively).42,46,47 Class I PI3K is the best-characterized family and the one most clearly implicated in human cancer,48 although much remains to be learned about their coupling to upstream signals and their relative functional output. In mammals, class I PI3Ks are present in all cell types, with p110γ and p110δ highly enriched in leukocytes.49 p110 subunits were originally divided into a class IA group (p110α, p110β, and p110δ), which bind the p85 type of regulatory subunit, and a class IB group (p110γ), which do not. Instead, p110γ binds 1 of 2 related regulatory subunits, p101 and p87, which have no homology to other proteins or recognizable domain structure.
Class II PI3Ks were discovered on the basis of their sequence homology with class I and class III PI3Ks rather than in a functional context. Mammals have 3 known members, PI3K-C2α (also known as PIK3C2A), PI3K-C2β (also PIK3C2B), and PI3K-C2γ (also known as PIK3C2G), but little is known about its role in signal transduction or in any other physiological mechanism. PI3K-C2α and PI3K-C2β have a broad but not ubiquitous tissue distribution, whereas the expression pattern of PI3K-C2γ seems to be more restricted.49 Finally, class III PI3Ks have only one known member, vacuolar protein sorting 34 or VPS34. Signals feeding into VPS34 are becoming apparent, but its physiological importance remains still unclear.47
Activation of PI3K can occur by at least 3 independent pathways, all of which start with binding of a ligand to receptor tyrosine kinases (RTKs; see Figure 1). This provokes the dimerization and autophosphorylation at tyrosine residues of the RTKs, which allows them to interact with Src homology 2 (SH2) domain–containing molecules.50,51 The first way to activate PI3K consists of the direct binding of the regulatory subunit of PI3K, p85, to phospho-YXXM motifs (in which X indicates any amino acid) within the RTK,52 triggering activation of the p110 catalytic subunit of PI3K. Other PI3K activation pathways depend on the adaptor protein GRB2, which binds preferentially to phospho-YXN motifs of the RTK.53 GRB2 binds to the scaffolding protein GAB, which in turn can bind to p85. Finally, the third way to activate PI3K pathways is via RAS. GRB2 binds and activates SOS, which then activates RAS, and this activates p110 independently of p85. GRB2 can also exist in a large complex that contains SOS, RAS, and GAB or other scaffolding proteins, bringing these activators into close proximity with p110 PI3K.54 It is not clear which of these pathways predominates in different physiological situations. Additionally, the p110β catalytic subunit can be activated by G protein–coupled receptors.42
Figure 1.
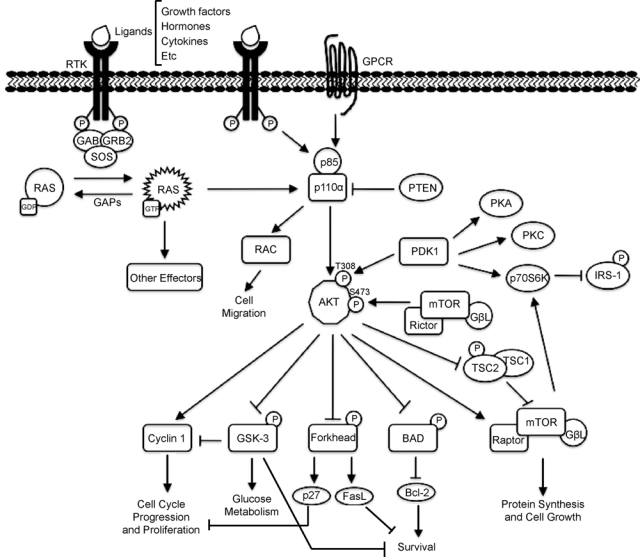
RAS signaling to the PI3K pathway.
The activation of PI3K results in the generation of the second messenger, phosphatidylinositol 3,4,5-triphosphate (PIP3) from phosphatidylinositol 4,5-bisphosphate (PIP2). The activation of PI3K and subsequent production of PIP3 drive the various downstream pathways that regulate a number of cellular functions including those involved in tumor development and progression. The tumor suppressor phosphatase and tensing homolog (PTEN) dephosphorylates PIP3 to PIP2, thereby terminating PI3K-dependent signaling. PIP3 propagates intracellular signaling by directly binding to proteins with PH domains,34 such as phosphoinositide-dependent kinase 1 (PDK1) and AKT. PIP3 brings PDK1 and AKT to the membrane, where PDK1 activates AKT by phosphorylation at residue threonine 308.55-57 PI3K-AKT signaling promotes cell growth and survival by several mechanisms. AKT promotes cell survival by inhibiting proapoptotic Bcl-2 family members BAD and BAX.34,46 AKT negatively regulates the transcription factor NF-κB, leading to an increase in transcription of antiapoptotic and prosurvival genes.58 AKT also phosphorylates Mdm2, and this activation antagonizes p53-mediated apoptosis. Furthermore, AKT negatively regulates forkhead transcription factors, thus reducing production of cell death–promoting proteins.58 AKT can also phosphorylate TSC2, thereby inhibiting the Rheb GTPase activity of the TSC1/ TSC2 dimer. Activated Rheb stimulates the mammalian target of rapamycin (mTOR), which contains protein complex mTORC1, leading to increased p70 S6 kinase activity. Activation of mTORC1 results in an increase in protein synthesis by phosphorylation of the eukaryotic initiation factor 4E and the ribosomal S6 protein.46 While mTORC1 relays signals following PI3K-AKT activation, a second mTOR complex, mTORC2, contributes to complete AKT activation by phosphorylating AKT on serine 473.59-61 Activation of the mTORC1 target, S6 kinase, negatively feeds back to decrease PI3K activation.
Several studies have established the central role of PI3K signaling in numerous cellular processes critical for cancer progression, including metabolism, growth, survival, and motility. Aberrant PI3K signaling is one of the most frequent occurrences in human cancer.46,62 Gene amplifications, deletions, and somatic missense mutation in the PIK3CA gene have been reported in many human cancer types, including cancers of the colon, breast, brain, liver, stomach, and lung. These somatic missense mutations were proposed to increase the kinase activity of PIK3CA, contributing to cellular transformation.63 Breast, endometrial, bladder, and colorectal carcinomas have been found to have a high rate of PIK3CA gene mutations, with frequencies of 26%, 22%, 21%, and 12%, respectively (COSMIC database: www.sanger.ac.uk/genetics/CGP/cosmic/). There appear to be 3 hotspot mutations within PIK3CA, H1047R, E542K, and E545K, corresponding to the helical and kinase domains of PI3K. Given the high degree of PI3K mutations in human cancers, significant efforts are being made to generate inhibitors for this pathway to treat cancers. Small molecule inhibitors of PI3K have entered into clinical trials in the past few years,64 but it will be some time before clear indications of efficacy against human cancer can be proven. Large bodies of preclinical data have been obtained with these agents during these years, including data from xenografts of human tumors in immunodeficient mice. Some of these studies have been encouraging, but until now, their effects have not been tested in animal models that could be considered to approximate to the human disease.
Properties of RAS-PI3K Interaction
The first evidence of an interaction between RAS and PI3K was obtained by the observation that in immunoprecipitates from RAS transformed cells, some PI3K activity could be found.65 Soon after that, it was described that RAS immobilized on agarose beads could bind to p110α/p85 when RAS was in its GTP-bound form and demonstrated that the level of PI3K products generated in vivo could be increased or decreased depending on whether activated or dominant-negative mutant RAS proteins were expressed.66 In the same report, it was also described that RAS interacts with PI3K in a direct manner without involving any other proteins, pointing to PI3K as an effector of RAS.66 Similar results were subsequently found using the fission yeast Schizosaccharomyces pombe as a model.67 Later on, it was shown that lysine residue 227 is essential for the interaction of RAS with PI3K and that RAS activates the Rho family GTPase Rac in a PI3K-dependent manner that is required for efficient transformation of fibroblasts by RAS.68
Initial crystallization trials of the RAS-PI3K interaction were unsuccessful. It was not until 2000 that the crystal structure of the RAS-PI3Kγ complex was first described.69,70 The structure of the RBD in PI3K consists of 5-stranded mixed β-sheets (Rβ1-Rβ5), flanked by 2 α-helices (Rα1 and Rα2). All RAS complexes with RBDs from different effectors described so far use a similar model of RAS effector interaction in which a β-sheet in the RAS and a β-sheet in the RBD are aligned to form a single β-sheet connecting the 2 proteins. Contacts between the switch I region of RAS and the RBD stabilize the interaction and ensure its dependence on RAS-GTP. PI3K, RAF, and RalGDS interact with many of the same switch I residues of RAS.71,72 In the structure of the RAS-PI3Kγ complex, contacts are primarily made via the switch I region of RAS and the RBD of PI3Kγ. However, a unique feature of the RAS-PI3Kγ complex is that the helix Rα1 and the Rα1-Rβ3 loop are significantly longer, and as a consequence, it causes a significant rotation of RAS relative to the RBD in PI3Kγ, resulting in essential intermolecular contacts with the RBD, thereby revealing the structural specificity of the RAS-PI3Kγ interaction.69,73 Uniquely for RAS interactions with effectors, there is contact between RAS and the catalytic domain of PI3Kγ, with R73 in the switch II region of RAS contacting E919 in the C-terminal lobe of the catalytic domain of PI3Kγ. This may contribute to the allosteric regulation of PI3K lipid kinase activity by RAS.
RAS is able to interact with the different isoforms of class I PI3Ks.66,68,74,75 For example, it was found that active RAS also binds to and activates p110γ catalytic subunit, provoking an approximately 8- to 20-fold increase in PI3Kγ activity. This activation is strictly dependent on RAS-GTP and is at least as great as the stimulation of the p110α/p85 heterodimer by RAS.68,69 The activating interaction of PI3Kγ with RAS is transient and limited by the dissociation of the RAS effector complex and not by the catalytic cycle of the effector enzyme. Direct interaction between GTP-bound RAS and p110α augments the activity of p110α, possibly by inducing a conformational change at the substrate binding site or by mediating a closer interaction with the plasma membrane.69,76 Indeed, the activity of p110γ that is defective in RAS binding can be restored with a myristoylation signal.
As previously mentioned in this review, activation by RAS is just one of the pathways contributing to PI3K activation. Binding of a ligand to RTKs provokes dimerization of the receptor and autophosphorylation at tyrosine residues. This allows them to interact with SH2 domain–containing proteins,50,51 such as GRB2, which binds and activates SOS (Son of Sevenless), and this, in turn, activates RAS. Thus, p110 is activated by RAS independently of p85. GRB2 also exists in a complex with GAB or other scaffolding proteins that interact with p85, bringing these activators into close proximity with p110 PI3K.54 There are data supporting the hypothesis that to activate p110, RAS has to function together with phosphotyrosine-bound p85. For example, HRAS promotes the catalytic activity of PI3K only when p85 is bound to phosphorylated tyrosine residues.77 So, RAS-mediated PI3K activation in response to growth factors requires 2 steps, the first of which is phosphorylation of the RTK and, in some cases, adaptor proteins, and secondly, activation of RAS small GTPases. The nature of the catalytic activation of PI3K by RAS has been a theme of debate, as it is difficult to distinguish between the contribution of membrane localization and RAS-induced allosteric lipid-kinase activation.
RAS-PI3K Signaling in Physiological Conditions
PI3Ks are one of the main effector families of RAS signaling. When active, PI3Ks convert phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2) and phosphatidylinositol (4)-phosphate (PI(4)P) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3) and phosphatidylinositol (3,4)-bisphosphate (PI(3,4)P2), respectively. The PIP3 produced will provoke accumulation and binding of signaling proteins with PH domains. The lipid products act in different pathways controlling mitogenic signaling, inhibition of apoptosis, intracellular vesicle trafficking and secretion, regulation of actin and integrin functions, and metabolic changes, often through 2 different protein kinases: Akt (also called protein kinase B or PKB) and p70 ribosomal protein S6 kinase (p70S6K). The lipid second-messenger molecules produced (PIP3 and PI(3,4)P2) activate the phosphoinositide-dependent kinases PDK1 and PDK2, which then activate Akt/PKB and isoforms of protein kinase C.13,34,78 The p70S6K route participates in protein biosynthesis, whereas Akt controls signaling pathways regulating different cellular processes such as cell survival, glucose uptake, and glycogen metabolism.13 PI3K can also activate the Rac GTPase, and this Rho family protein is an important mediator of oncogenic RAS transformation. In fact, RAS and PI3K activities are necessary for Rac activation through the exchangers Sos and Vav.79,80 The signaling pathway is inactivated by the action of the tumor suppressor PTEN, which antagonizes PI3K activity via its intrinsic lipid phosphatase activity, thus reducing the cellular pool of PIP3 by converting it back to PI(4,5)P2.81-83
The first developmental link between RAS and PI3K was established in Drosophila.84 More recently, it was shown that direct interaction of PI3K with RAS was critical in this setting.85 Transgenic flies carrying a Dp110 (the only Drosophila p110) with mutations in the RBD that did not interact with RAS but were otherwise biochemically normal were generated. In this model, it was shown that Dp110 mutant flies were viable, thus establishing that RAS-mediated Dp110 regulation is dispensable for viability. However, the survivor flies were smaller in size, and the females presented a decreased fecundity, laying around 60% fewer eggs. Dp110 mutant flies also had decreased activation of Akt in response to insulin in the brain and imaginal discs and decreased basal Akt activation in the ovaries. This work demonstrated for the first time that the interaction between RAS and PI3K is essential in specific developmental circumstances, requiring maximal PI3K signaling.
Most of the initial knowledge about the interaction between mammalian PI3K and RAS has been acquired using purified or overexpressed proteins in vitro and in cultured cells. Suire et al. were the first describing the in vivo role of RAS-PI3K interaction in mammals.86 They generated a mouse model in which the RBD of p110γ (p110γDASAA) was mutated; the resulting mice were viable, fertile, of normal size, and with generally normal blood counts. However, in isolated neutrophils, production of PI(3,4)P2 and PIP3, activation of Akt, and chemotaxis were decreased compared with wild-type cells or cells from p110γ heterozygous mice. The production of reactive oxygen species in response to agonist stimulation was decreased in p110γ-RBD mutant mice, revealing an important role for the interaction in the normal function of neutrophils.
Further evidence of the importance of the RAS-PI3K interaction in vivo was provided by the generation of a knockin mouse having 2 point mutations in the RBD of p110α (T208D and K227A). By the insertion of these mutations, the interaction between RAS and p110α was completely disrupted, but it did not affect the enzymatic activity of p110α.87 This mouse model showed some perinatal lethality in homozygotes, with the survivors being smaller in size than their wild-type counterparts. Furthermore, they exhibited defective branching and development of the lymphatic vasculature system. This defect was linked with the development of chylous ascites in the newborn mice and is similar to developmental anomalies seen in mice with defective VEGF-C signaling.88 In cultured mouse embryo fibroblasts, loss of p110α binding to RAS strongly reduces Akt activation in response to certain growth factors such as EGF or FGF2.
RAS-dependent PI3K activation is linked to prosurvival signaling due to the activation of Akt. Studies showing that activated PI3K or Akt abrogates apoptosis and that dominant-negative Akt has the ability to enhance apoptosis have highlighted the importance of PI3K/Akt signaling to promote survival.32,89-91 Akt phosphorylates a number of substrates that play a role in the regulation of apoptosis. For example, Akt phosphorylation of Bad, a proapoptotic member of the Bcl-2 family of apoptotic regulators, causes Bad to bind preferentially to 14-3-3 in an inactive complex, thereby preventing it from sequestering and inactivating the antiapoptotic proteins Bcl-2 and Bcl-XL.89,92,93 Also, Akt and Rac facilitate RAS activation of the NF-κB transcription factor,94-96 which serves an antiapoptotic role in RAS function.97 Moreover, PI3K is absolutely required for the proliferative response to RAS in human thyroid epithelial cells, acting via suppression of RAS-induced apoptosis.92 Akt also phosphorylates the FOXO family of transcription factors.98 Thus, by increasing the ability for growth and by decreasing the capacity for apoptosis, PI3K signaling supports tumorigenesis.
It is well established that PI3Ks are one of the principal effectors of RAS signaling to the cell cycle control machinery.99,100 RAS activity is needed throughout all the different phases of cell cycle. In quiescent cells stimulated with growth factors, PI3K has been shown to be activated twice, as cells transition from G0 into G1 phase99,101,102 and then later in G1 phase. However, the use of PI3K inhibitors has established that it is only during the later stages of G1 phase that PI3K activity promotes entry into S phase.99
Use of KRAS knockout fetal liver cells has shown that KRAS signals to PI3K to regulate differentiation and proliferation of erythroid progenitor cells.103,104 Erythropoietin (EPO) and EPO receptor have been found to be able to activate RAS-MAPK and PI3K pathways.105 Studies on human erythroid progenitors showed the importance of PI3K for RAS, MEK, and ERK activation, which were stimulated by EPO through a RAF-independent way.106 All these suggest the important involvement of RAS signaling in hematopoiesis and highlight the importance and complexity of PI3K-Akt and/or RAF-MAPK in connecting to and mediating RAS signaling. Using zebrafish as a model, it has been shown that PI3K-Akt signaling pathway is a key regulator of RAS signaling during both hematopoiesis and angiogenesis.107 Taking all these data together, they suggest that there might exist a potential crosstalk between PI3K and MAPK pathways, such as the studies in hematopoietic progenitor, which demonstrated that MAPK pathway through RAS is PI3K dependent and that PI3K drives RAF/MEK/ERK activation through RAF by a yet uncharacterized mechanism.108
Importance of the RAS-PI3K Interaction during Oncogenic Signaling
Although the importance of RAS proteins in developmental processes is generally appreciated, it is the oncogenic potential of constitutively activated RAS proteins that has attracted most attention. The implication of PI3K as an important target in RAS-dependent transformation was established soon after the discovery of the interaction of these 2 molecules.68,109-111
RAS signaling pathways are activated in tumorigenesis by a number of different mechanisms, including mutations in RAS, loss of GAP proteins, overexpression of RTKs (EGFR, ERBB2), and also mutations or amplifications in any of their effector molecules.32,112-114 In recent years, it has become evident that many human tumors harbor somatic missense mutations in PIK3CA at high frequency, including tumors of the brain, breast, colon, liver, stomach, lung, and ovary.62,115-120 Particularly high incidences of mutation are found in cancers of the breast, the colon, or the endometrium (catalog of Somatic Mutations in Cancer: http://www.sanger.ac.uk/genetics/CGP/cosmic). These are point mutations that in about 80% of the cases map to 3 hotspots in the coding sequence of PIK3CA. Two of the hotspots, represented by the single amino acid substitutions E542K and E545K, are localized in the helical domain of the protein; the third, represented by the H1047R substitution, resides in the kinase domain. It has been shown in different systems that these are oncogenic gain-of-function mutations that increase enzymatic activity, constitutively stimulate Akt signaling, induce growth factor– and anchorage-independent growth in culture, and cause tumors in vivo.121-127 Recently, it has been demonstrated that helical and kinase domain mutations trigger gain of function through different mechanisms, showing a differential requirement for interaction with the regulatory subunit p85 and with RAS-GTP. The gain of function seen with helical domain mutations depends on an interaction with RAS-GTP but is less affected by binding to p85. In contrast, the kinase domain mutation is active in the absence of RAS-GTP binding but is dependent on allosteric change mediated by p85, and this allosteric change mimics RAS-GTP binding.128-130
One of the clearest examples showing the importance of the RAS-PI3K interaction in RAS-driven tumorigenesis was provided by the above-mentioned RBD-p110α mutant knockin model. First, mice homozygous for mutant PIK3CA were crossed with the K-RAS LA2 mice,131 and in this model, an impressive 95% reduction in lung tumor formation was found. Second, when PIK3CA mutant mice were subjected to DMBA-induced carcinogenesis, a near-complete reduction in H-RAS–driven tumor formation was seen.87 The use of these 2 well-characterized mouse models of endogenous RAS-driven tumorigenesis demonstrated that p110α is a critical effector for RAS-driven tumorigenesis, at least in lung and skin tumors.
Whereas RAS and PI3K pathway mutations are found to be mutually exclusive in breast cancers, tumors with mutant p110α often coexist with mutations in RAS in colorectal and endometrial cancer.18,125,132,133 The molecular significance and therapeutic implications of co-occurring mutations are unclear, but it could be argued that additional mutations enhance the oncogenic transformation by strengthening PI3K pathway signaling caused by oncogenic RAS, thus activating other pathways. The use of inhibitors and shRNA studies have shown that signaling to downstream molecules such as AKT is mediated by p110α in colon and endometrial cancers with PIK3CA and coexisting RAS mutations.132 In agreement, in HCT116 and DLD1 colon cancer cell lines harboring mutant p110α and KRAS, somatic cell knockout of p110α inhibits downstream PI3K signaling, cell growth and transformation, and maintenance of tumor xenografts.125
There is increasing evidence that cholesterol content may be a critical determinant of tumor progression. Both their content and biosynthesis are increased in different proliferating tumoral cells compared with normal cells, including colorectal cancer cells.134,135 In this regard, it has been shown that oncogenic RAS, but not oncogenic BRAF, protects from apoptosis induced by cholesterol depletion through JNK activation in colorectal cancer cells and that the protective role of oncogenic RAS is exerted through constitutive activation of the PI3K/AKT pathway. Expression of oncogenic RAS or constitutively activated AKT significantly decreases the cholesterol depletion–induced apoptosis in HT-29, whereas abrogation of AKT expression in HCT-116 cells renders these cells sensitive to apoptosis by cholesterol depletion.136 Thus, strategies that induce apoptosis selectively in cancer cells, such as cholesterol depletion, together with PI3K inhibitors should greatly increase the chemopreventive effects or allow their use at lower doses and are therefore highly desirable.
Data from different studies suggest that, apart from p110α, RAS also needs p110β interaction in some occasions to exert its oncogenic role. Thus, MEFs lacking p110α are resistant to oncogenic RAS transformation,137 and in some human breast cancer cell lines that express an oncogenic HRAS but carry wild-type PIK3CA and PTEN, inhibitors of p110α, but not p110β, block PI3K signaling without affecting cell proliferation.138 In some other cases, RAS-p110β interaction has been shown to be essential for RAS-driven tumorigenesis because ablation of this isoform completely abrogates focus formation induced by oncogenic HRAS.139
Tumor cells become addicted to the expression of initiating oncogenes, such that loss of oncogene expression in established tumors leads to tumor regression.140 It is well accepted that oncogenic RAS promotes both initiation and maintenance of tumor growth.141 Considering the vast array of RAS effectors, it is important to address what is the specific involvement of each of them in RAS tumor maintenance. In this regard, it has been shown that PI3K signaling is indispensable to maintain transformed growth in RAS mutant cell lines both in vitro and in xenografts in mice. Although multiple RAS effectors are essential to initiate tumor formation, only signaling through the PI3K/Akt pathway seems to be necessary to maintain tumor growth.142 The reduction of RAS oncogene dependence to activation of PI3K/Akt pathway appears to be a consequence of redundant signaling provided by the established tumor microenvironment. It has also been described that the requirement for PI3K signaling during tumor maintenance in RAS-driven tumor growth might be due, at least in part, to the Akt-dependent activation of eNOS (by phosphorylation in S1177). This, in turn, leads to S-nitrosylation of normal RAS proteins, activating them perhaps as a means to diversify the signal beyond that of oncogenic RAS.143
While the data above suggest that PI3K signaling is essential for RAS-driven tumor maintenance, recent data have suggested that inhibition of PI3K alone is not sufficient to cause regression of tumors once established. When mice with genetic deletion of the p85 PI3K regulatory subunits were crossed with a Tet-inducible K-rasG12D transgenic model144 or with the LSL K-RAS model,145 the induction of lung tumor formation was reduced relative to a PI3K wild-type background. However, the lung tumors that developed in response to KRAS activation in mice with normal PI3K signaling failed to reduce in size when these animals were treated with NVP-BEZ235, a dual pan-PI3K/mTOR inhibitor, even though tumors driven by expression of activated PI3K did regress with this drug.146 However, when these tumors were treated with the PI3K inhibitor combined with a MEK inhibitor, they underwent an impressive regression, suggesting that a combination of PI3K and MEK inhibitors may have potential for the treatment of RAS mutant tumors in the clinic, provided, of course, that toxicities are not limiting.
Much of our knowledge of human cell transformation derives from studies undertaken in rodent cells. However, it must be taken into account that there exist fundamental differences in the behavior of rodent and human cells, especially in certain biological features relevant to transformation.147-149 In this regard, RAS-dependent transformation presents differences in the effector pathway needed for effective transformation not only between mouse and human cell lines but also when comparing human cell lines originating from different tissue types.148 While the ab initio creation of tumor cell lines from normal cells in vitro that are used in these studies has provided many insights into the process of tumorigenesis, it is still unclear exactly how closely this correlates with the processes occurring during the normal pathological process of human tumor formation in vivo.
RAS-PI3K in Metastasis Formation
Formation of metastases is the leading cause of death in cancer patients. Metastasis establishment is a multistep process that involves dissociation of cancer cells from the primary tumor, invasion of extracellular matrix, angiogenesis, intravasation into the vasculature or lymphatic systems, survival in these places, extravasation, and proliferation at a distant site.150-153 Oncogenic RAS signaling facilitates almost all aspects of malignant phenotype.154,155 However, it is still unclear the specific role it plays in tumor invasion and metastasis or the main RAS effector pathway that contributes to metastasis formation, although several studies suggest that PI3K is exerting a key role in it. In addition to work in mammalian cells, many insights into the role of RAS and PI3K in cell motility, and especially chemotaxis, have also come from the study of the slime mold Dictyostelium discoideum.156
In order to acquire a migratory phenotype, tumoral cells must undergo extensive actin cytoskeleton remodeling, which is in part regulated by Rho family GTPases (including RhoA, Rac1, and Cdc42). The ability of activated RAS to stimulate PI3K in addition to RAF is important in RAS transformation of mammalian cells and essential in RAS-induced cytoskeletal reorganization.109 RAS activates Rac via PI3K, and actin rearrangement correlates with the ability of RAS mutants to activate PI3K. Inhibition of PI3K activity blocks RAS induction of membrane ruffling, while activated PI3K is sufficient to induce membrane ruffling, acting through Rac. How RAS regulates RhoA and Cdc42 function is still not clearly understood. PI3K can also activate Rac GEFs such as Sos or Vav to promote activation of Rac.79,80 Rac regulation of actin reorganization and membrane ruffling can promote increased cell motility and contribute to tumor cell invasion and metastasis.157
An interesting connection between PI3K signaling and RAS-induced malignant transformation involves the small GTPase RhoB, which is a potent suppressor of transformation, migration, and invasion of different cancer cells and NIH3T3 fibroblasts.158 Expression of RhoB is downregulated by oncogenic HRAS dependent on PI3K but not RAF signaling pathways. In contrast, ectopic expression of RhoB antagonizes RAS-PI3K–dependent malignant transformation and apoptosis.
The changes in cell-cell adhesion and cell-ECM interaction that are regulated by PI3K signaling might cause important consequences for the regulation of cell motility. In uveal melanoma, PI3K pathway downregulates the cell adhesion molecules E-cadherin and β-catenin, thus attenuating cell-cell adhesion and promoting the enhanced motility and migration typically seen in these cancer cells.159 Also, in MDA-MB-435 breast carcinoma cells, upregulation of PI3K via α6β4 integrin signaling leads to increased invasion that is Rac dependent but MAPK and p70S6K independent.160,161 β4 integrin contains Shc motifs that can recruit Grb2 and Sos, so it can be speculated that RAS activation might be responsible for this increase in migration. It has been demonstrated that PI3K activation can enhance invasion by regulating expression of different matrix metalloproteases such as MMP-9 in HT1080 cells162 or MMP-2 in mouse mammary epithelial cells.163 However, whether these mechanisms are directly related to RAS regulation of PI3K is not clear.
The nonreceptor tyrosine kinase focal adhesion kinase (FAK) is a major mediator of integrin signaling.164 Engagement of β1 and αv integrins induces FAK localization to matrix adhesions and activations. Through a plethora of effectors, FAK regulates focal adhesion dynamics during cell migration and activates prosurvival and mitogenic signaling pathways.165 Genetic studies have indicated that FAK regulates cell survival and cell proliferation in certain cell types.166,167 Furthermore, several observations suggest that FAK is not required for neoplastic transformation but promotes cancer cell invasion,165 and a large fraction of breast cancers express elevated levels of FAK.168,169 Recently, it has been shown that in a model of breast cancer, FAK supports RAS- and PI3K-dependent neoplastic transformation by orchestrating multiple core functions, including proliferation, survival, and avoidance of senescence. In addition, FAK is necessary for tumor invasion and metastasis.170 This dependency of RAS- and PI3K-transformed mammary tumor cells on FAK signaling has broad biological implications and identifies a vulnerability that could be exploited therapeutically.
Oncogenic RAS and PI3K can promote the loss of anchorage-dependent growth, which is a key feature needed for tumor cells to form metastases.171-173 In MDCK epithelial cells, the PI3K pathway, but not RAF pathway, is both necessary and sufficient for the protection provided by RAS from anoikis (or matrix deprivation–induced apoptosis).174,175 This inhibition of anoikis gives the detached tumor cell the possibility to migrate to a conduit by which it can reach distal tissues.
Although much work has been accomplished in discovering the contribution of oncogenic RAS to increased motility, invasiveness, and metastatic potential, mechanisms downstream of RAS are much more complex than originally thought. There are still many facets of the contribution of PI3K to oncogenic RAS invasiveness and metastatic potential that remain unanswered. Appropriate knowledge of this contribution might be critical for the development of proper therapy against metastasis.
Interconnections with Other RAS-Regulated Pathways
It is known that PI3K and MAPK pathways can interact in multiple ways (see Figure 2), both in normal and transforming conditions.109,176 Crosstalk regulation between RAF-MAPK and PI3K-AKT pathways was first described during muscle cell differentiation,177,178 although in this system, both pathways exert opposing effects. PI3K-Akt inhibits the RAF pathway in muscle cell hypertrophy depending on the differentiation state of the cell: at the stage of differentiated myotubes, Akt activation inhibits the RAF-MEK-Erk pathway; however, Akt activation has no effect over Erk pathway in their myoblast precursors.177
Figure 2.
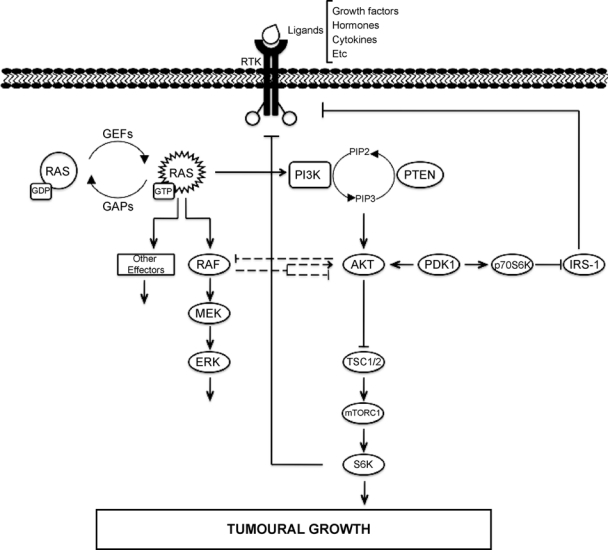
Crosstalk and feedback in ERK and PI3K signaling downstream of RAS.
Another level of interconnection between these 2 RAS-regulated pathways can be observed at the level of RAF. The PI3K inhibitor LY294002 and the downstream p70S6K inhibitor rapamycin are potent inhibitors of RAF-mediated proliferation, suggesting that the PI3K/Akt/p70S6K pathway mediates some effects of RAF on cell growth.176 Akt interacts with and phosphorylates RAF protein within its regulatory domain at a highly conserved serine residue (Ser259). This phosphorylation inhibits activation of the RAF signaling pathway and shifts the cellular response in a human breast cancer cell line from cell cycle arrest to proliferation.178 It has also been established that constitutive RAF-MEK1 signaling leads to negative feedback inhibition of RAS and PI3K through the Ephrin receptor EphA2 and that this event is required for cellular arrest.179 PI3K inhibition of RAF-MAPK signaling has been shown to be also required for vascular smooth muscle and intestinal epithelial cell differentiation.180,181 However, RAF-MAPK signaling can be a potent inducer of both RAS and PI3K activation, especially in epithelial cells, through the autocrine production of growth factors such as HB-EGF.182 Overall, the crosstalk between the PI3K and RAF pathways is made up of a complex network of events, some of which are likely to be more significant than others under physiological signaling conditions. It has also been described that in human pancreatic duct epithelial cells, inactivation of the MAPK pathway using different inhibitors decreases Akt phosphorylation,191 suggesting that the interaction between Raf/MEK/ERK and PI3K/Akt pathways is important in RAS-mediated transformation of human cells.
The mTOR (mammalian target of rapamycin) kinase can also connect these 2 pathways in a RAS-dependent manner.183 mTOR can be found in 2 different complexes, mTORC1 (containing RAPTOR) and mTORC2 (containing RICTOR). Both PI3K and MAPK signaling pathways regulate mTORC1 activity through phosphorylation of the tuberous sclerosis complex 2 (TSC2).184-187 In turn, the mTORC1 target p70S6K phosphorylates the adaptor protein IRS1, leading to inhibition of insulin signaling to PI3K and Akt,188-190 thus forming a negative feedback loop. Additionally, it has been described that inhibition of mTORC1 by RAD001 in patients with metastatic disease leads to an activation of the MAPK pathway.183 The authors proposed that mTORC1 inhibition increases RTK activity toward RAS/MAPK, thus promoting AKT and ERK activation in a dual feedback mechanism.
Development of Inhibitors
The fact that RAS genes are the most mutated oncogenes in human cancer makes the proteins they encode and the signaling pathways that they regulate attractive targets for drug development. The greatest pharmaceutical effort made to abolish RAS oncogenic signaling was the development of drugs targeting farnesyltransferases, which were thought to be essential for RAS biological activity because they regulate their binding to biological membranes. Although many drugs were successfully developed that inhibited farnesyltransferase, they failed in human patient studies principally due to the ability of KRAS and NRAS to use an alternative modification enzyme (geranylgeranyltransferase) to localize to membranes.192
A more successful attempt to target RAS signaling pathways has been the development of inhibitors for RTKs.193 However, the use of these compounds is restricted to patients presenting oncogenic RTK signaling, while patients with tumors expressing an oncogenic mutant form of RAS do not benefit from such compounds because oncogenic RAS acts downstream to circumvent the need for an oncogenic RTK to induce cell proliferation and survival.194
More recent therapeutic attempts have focused on the inhibition of PI3Ks. The fact that PI3K activation might have an important role during tumor maintenance highlights the importance of this pathway as an anticancer target.142,143 Because cancers are treated at the tumor maintenance stage, targeting this aspect of RAS oncogenesis may hold promise as an approach to the management of RAS-driven human cancers. In this regard, the mouse model generated in our laboratory in which the RAS-PI3K interaction has been disrupted by mutations in the RBD of p110α87 provides helpful insight into the benefits that might be achieved from therapeutic compounds developed to target the RAS-PI3K interaction in human tumors. The development of such a drug might improve the condition of cancer patients in which the oncogenicity of RAS is exerted via PI3K pathway and, as a consequence of the specificity of the interaction targeted, might minimize the side effects in the patients, one of the main prerequisites for all novel therapeutic compounds. It would also overcome the feedback loops that exist between MAPK and PI3K pathways.
It might also be considered that p110α and p110β might be activated in different types of cancer. Although the role of oncogenic RAS signaling through p110β is not very well established, both p110α and p110β might be useful drug targets in particular tumor types bearing mutant RAS proteins. Hence, compounds targeting the specific isoform might be a good option in tumors featuring these lesions.114 It can also be speculated that the differences found in the mutations in the kinase and helical domains of p110α might induce conformational rearrangements that are unique and of sufficient magnitude to be exploitable for the identification of small molecule inhibitors that are mutant specific but do not affect the WT protein. Because of their exclusive effects on cancer tissue, such inhibitors could have therapeutic properties that are superior to those of pan-specific PI3K inhibitors.
Because RAS coordinately activates both the PI3K and RAF/MEK/MAPK signaling pathways, it seems very attractive to use a combination of MAPK pathway (either MEK, pan-RAF, or BRAF) and PI3K inhibitors in the treatment of RAS-driven cancers. As well as recent data discussed above favoring the use of a combination of PI3K and MEK inhibition to hit established RAS-induced tumors,146 it has also been shown that activation of the PI3K pathway, either by PIK3CA mutations or PTEN loss, is a major resistance mechanism that impairs the efficiency of MEK inhibitors in KRAS mutated cancers. Recently, it has been shown that in RAS mutant tumors, PIK3CA mutations restore cyclin D1 expression and G1-S cell cycle progression so that they are no longer dependent on RAS and MEK/ERK signaling.195 Downregulation of p110α resensitizes tumors with KRAS and PIK3CA mutations to MEK pathway inhibition both in vitro and in vivo.196 This might be an explanation of why RAS mutant cancers exhibit a variable response to MEK inhibitors.197 Similar results regarding the clinical benefits of downregulation of both pathways have been shown in a mouse model of prostate cancer198 and in thyroid tumor cells,199 and in hematopoietic cell lines, it has been proven that the combined inhibition of MEK and PI3K pathways provokes a stronger effect in apoptosis induction and growth inhibition than single pathway inhibition.200 Taken together, these data provide a strong rationale for the combination of PI3K and MEK inhibitors in cancers having an oncogenic KRAS mutation.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature. 1990;348:125-32 [DOI] [PubMed] [Google Scholar]
- Field J, Broek D, Kataoka T, Wigler M. Guanine nucleotide activation of, and competition between, RAS proteins from Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:2128-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Nakamura S, Kaziro Y. Induction of neurite formation in PC12 cells by microinjection of proto-oncogenic Ha-ras protein preincubated with guanosine-5’-O-(3-thiotriphosphate). Mol Cell Biol. 1987;7:4553-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittinghofer A, Pai EF. The structure of Ras protein: a model for a universal molecular switch. Trends Biochem Sci. 1991;16:382-7 [DOI] [PubMed] [Google Scholar]
- Wolfman A, Macara IG. A cytosolic protein catalyzes the release of GDP from p21ras. Science. 1990;248:67-9 [DOI] [PubMed] [Google Scholar]
- Trahey M, McCormick F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science. 1987;238:542-5 [DOI] [PubMed] [Google Scholar]
- Hirakawa T, Ruley HE. Rescue of cells from ras oncogene-induced growth arrest by a second, complementing, oncogene. Proc Natl Acad Sci U S A. 1988;85:1519-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596-602 [DOI] [PubMed] [Google Scholar]
- Ruley HE. Adenovirus early region 1A enables viral and cellular transforming genes to transform primary cells in culture. Nature. 1983;304:602-6 [DOI] [PubMed] [Google Scholar]
- Land H, Parada LF, Weinberg RA. Cellular oncogenes and multistep carcinogenesis. Science. 1983;222:771-8 [DOI] [PubMed] [Google Scholar]
- Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682-9 [PubMed] [Google Scholar]
- Repasky GA, Chenette EJ, Der CJ. Renewing the conspiracy theory debate: does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004;14:639-47 [DOI] [PubMed] [Google Scholar]
- Rojas JM, Santos E. ras genes and human cancer: different implications and different roles. Curr Genom. 2002;3:295-311 [Google Scholar]
- Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593-604 [DOI] [PubMed] [Google Scholar]
- Shannon KM, O’Connell P, Martin GA, et al. Loss of the normal NF1 allele from the bone marrow of children with type 1 neurofibromatosis and malignant myeloid disorders. N Engl J Med. 1994;330:597-601 [DOI] [PubMed] [Google Scholar]
- Side L, Taylor B, Cayouette M, et al. Homozygous inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis type 1 and malignant myeloid disorders. N Engl J Med. 1997;336:1713-20 [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54 [DOI] [PubMed] [Google Scholar]
- Parsons DW, Wang TL, Samuels Y, et al. Colorectal cancer: mutations in a signalling pathway. Nature. 2005;436:792. [DOI] [PubMed] [Google Scholar]
- Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science. 1993;260:1658-61 [DOI] [PubMed] [Google Scholar]
- Warne PH, Viciana PR, Downward J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature. 1993;364:352-5 [DOI] [PubMed] [Google Scholar]
- Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74:205-14 [DOI] [PubMed] [Google Scholar]
- Zhang XF, Settleman J, Kyriakis JM, et al. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature. 1993;364:308-13 [DOI] [PubMed] [Google Scholar]
- Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian JR, Vojtek AB, Cooper JA, Morrison DK. A single amino acid change in Raf-1 inhibits Ras binding and alters Raf-1 function. Proc Natl Acad Sci U S A. 1994;91:5982-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais R, Light Y, Mason C, Paterson H, Olson MF, Marshall CJ. Requirement of Ras-GTP-Raf complexes for activation of Raf-1 by protein kinase C. Science. 1998;280:109-12 [DOI] [PubMed] [Google Scholar]
- Marais R, Light Y, Paterson HF, Marshall CJ. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995;14:3136-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison D. 14-3-3: modulators of signaling proteins? Science. 1994;266:56-7 [DOI] [PubMed] [Google Scholar]
- Avruch J, Khokhlatchev A, Kyriakis JM, et al. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog Horm Res. 2001;56:127-55 [DOI] [PubMed] [Google Scholar]
- Crews CM, Erikson RL. Extracellular signals and reversible protein phosphorylation: what to Mek of it all. Cell. 1993;74:215-7 [DOI] [PubMed] [Google Scholar]
- Rapp UR, Goldsborough MD, Mark GE, et al. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc Natl Acad Sci U S A. 1983;80:4218-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons JF, Wilhelm S, Hibner B, Bollag G. Discovery of a novel Raf kinase inhibitor. Endocr Relat Cancer. 2001;8:219-25 [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489-501 [DOI] [PubMed] [Google Scholar]
- Castellano E, Downward J. Role of RAS in the regulation of PI 3-kinase. Curr Top Microbiol Immunol. 2011;346:143-69 [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655-7 [DOI] [PubMed] [Google Scholar]
- D’Adamo DR, Novick S, Kahn JM, Leonardi P, Pellicer A. rsc: a novel oncogene with structural and functional homology with the gene family of exchange factors for Ral. Oncogene. 1997;14:1295-305 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, McCormick F. RalGDS comes of age. Cancer Cell. 2005;7:205-6 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia A, Pritchard CA, Paterson HF, Mavria G, Stamp G, Marshall CJ. RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell. 2005;7:219-26 [DOI] [PubMed] [Google Scholar]
- White MA, Vale T, Camonis JH, Schaefer E, Wigler MH. A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J Biol Chem. 1996;271:16439-42 [DOI] [PubMed] [Google Scholar]
- Ehrhardt A, Ehrhardt GR, Guo X, Schrader JW. Ras and relatives: job sharing and networking keep an old family together. Exp Hematol. 2002;30:1089-106 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Sabatier C, McCormick F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Mol Cell Biol. 2004;24:4943-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley LC, Auger KR, Carpenter C, et al. Oncogenes and signal transduction. Cell. 1991;64:281-302 [DOI] [PubMed] [Google Scholar]
- Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615-75 [DOI] [PubMed] [Google Scholar]
- Volinia S, Hiles I, Ormondroyd E, et al. Molecular cloning, cDNA sequence, and chromosomal localization of the human phosphatidylinositol 3-kinase p110 alpha (PIK3CA) gene. Genomics. 1994;24:472-7 [DOI] [PubMed] [Google Scholar]
- Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481-507 [DOI] [PubMed] [Google Scholar]
- Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606-19 [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329-41 [DOI] [PubMed] [Google Scholar]
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497-510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok K, Geering B, Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem Sci. 2009;34:115-27 [DOI] [PubMed] [Google Scholar]
- Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445-52 [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;110:669-72 [DOI] [PubMed] [Google Scholar]
- Domchek SM, Auger KR, Chatterjee S, Burke TR, Jr., Shoelson SE. Inhibition of SH2 domain/phosphoprotein association by a nonhydrolyzable phosphonopeptide. Biochemistry. 1992;31:9865-70 [DOI] [PubMed] [Google Scholar]
- Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191-203 [DOI] [PubMed] [Google Scholar]
- Ong SH, Hadari YR, Gotoh N, Guy GR, Schlessinger J, Lax I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci U S A. 2001;98:6074-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261-9 [DOI] [PubMed] [Google Scholar]
- Currie RA, Walker KS, Gray A, et al. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem J. 1999;337(Pt 3):575-83 [PMC free article] [PubMed] [Google Scholar]
- Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005;24:7465-74 [DOI] [PubMed] [Google Scholar]
- Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J. 2008;415:333-44 [DOI] [PubMed] [Google Scholar]
- Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280:40406-16 [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098-101 [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159-68 [DOI] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. [DOI] [PubMed] [Google Scholar]
- Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer. 2006;94:455-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784:159-85 [DOI] [PubMed] [Google Scholar]
- Sjolander A, Yamamoto K, Huber BE, Lapetina EG. Association of p21ras with phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 1991;88:7908-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Dhand R, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527-32 [DOI] [PubMed] [Google Scholar]
- Kodaki T, Woscholski R, Hallberg B, Rodriguez-Viciana P, Downward J, Parker PJ. The activation of phosphatidylinositol 3-kinase by Ras. Curr Biol. 1994;4:798-806 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996;15:2442-51 [PMC free article] [PubMed] [Google Scholar]
- Pacold ME, Suire S, Perisic O, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103:931-43 [DOI] [PubMed] [Google Scholar]
- Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313-20 [DOI] [PubMed] [Google Scholar]
- Nassar N, Horn G, Herrmann C, Scherer A, McCormick F, Wittinghofer A. The 2.2 A crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature. 1995;375:554-60 [DOI] [PubMed] [Google Scholar]
- Huang L, Hofer F, Martin GS, Kim SH. Structural basis for the interaction of Ras with Ral-GDS. Nat Struct Biol. 1998;5:422-6 [DOI] [PubMed] [Google Scholar]
- Djordjevic S, Driscoll PC. Structural insight into substrate specificity and regulatory mechanisms of phosphoinositide 3-kinases. Trends Biochem Sci. 2002;27:426-32 [DOI] [PubMed] [Google Scholar]
- Rubio I, Rodriguez-Viciana P, Downward J, Wetzker R. Interaction of Ras with phosphoinositide 3-kinase gamma. Biochem J. 1997;326(Pt 3):891-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Welham MJ, Kotani K, et al. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci U S A. 1997;94:4330-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denley A, Kang S, Karst U, Vogt PK. Oncogenic signaling of class I PI3K isoforms. Oncogene. 2008;27:2561-74 [DOI] [PubMed] [Google Scholar]
- Chan TO, Rodeck U, Chan AM, et al. Small GTPases and tyrosine kinases coregulate a molecular switch in the phosphoinositide 3-kinase regulatory subunit. Cancer Cell. 2002;1:181-91 [DOI] [PubMed] [Google Scholar]
- Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: ’it ain’t over ’til it’s over’. Trends Cell Biol. 2000;10:147-54 [DOI] [PubMed] [Google Scholar]
- Nimnual AS, Yatsula BA, Bar-Sagi D. Coupling of Ras and Rac guanosine triphosphatases through the Ras exchanger Sos. Science. 1998;279:560-3 [DOI] [PubMed] [Google Scholar]
- Han J, Luby-Phelps K, Das B, et al. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 1998;279:558-60 [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375-8 [DOI] [PubMed] [Google Scholar]
- Maehama T, Taylor GS, Dixon JE. PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem. 2001;70:247-79 [DOI] [PubMed] [Google Scholar]
- Wishart MJ, Dixon JE. PTEN and myotubularin phosphatases: from 3-phosphoinositide dephosphorylation to disease. Trends Cell Biol. 2002;12:579-85 [DOI] [PubMed] [Google Scholar]
- Prober DA, Edgar BA. Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes Dev. 2002;16:2286-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orme MH, Alrubaie S, Bradley GL, Walker CD, Leevers SJ. Input from Ras is required for maximal PI(3)K signalling in Drosophila. Nat Cell Biol. 2006;8:1298-302 [DOI] [PubMed] [Google Scholar]
- Suire S, Condliffe AM, Ferguson GJ, et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat Cell Biol. 2006;8:1303-9 [DOI] [PubMed] [Google Scholar]
- Gupta S, Ramjaun AR, Haiko P, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957-68 [DOI] [PubMed] [Google Scholar]
- Karkkainen MJ, Haiko P, Sainio K, et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. 2004;5:74-80 [DOI] [PubMed] [Google Scholar]
- Cox AD, Der CJ. The dark side of Ras: regulation of apoptosis. Oncogene. 2003;22:8999-9006 [DOI] [PubMed] [Google Scholar]
- Downward J. Ras signalling and apoptosis. Curr Opin Genet Dev. 1998;8:49-54 [DOI] [PubMed] [Google Scholar]
- Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15:177-82 [DOI] [PubMed] [Google Scholar]
- Gire V, Marshall C, Wynford-Thomas D. PI-3-kinase is an essential anti-apoptotic effector in the proliferative response of primary human epithelial cells to mutant RAS. Oncogene. 2000;19:2269-76 [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231-41 [DOI] [PubMed] [Google Scholar]
- Irani K, Xia Y, Zweier JL, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649-52 [DOI] [PubMed] [Google Scholar]
- Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86-90 [DOI] [PubMed] [Google Scholar]
- Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82-5 [DOI] [PubMed] [Google Scholar]
- Mayo MW, Wang CY, Cogswell PC, et al. Requirement of NF-kappaB activation to suppress p53-independent apoptosis induced by oncogenic Ras. Science. 1997;278:1812-5 [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857-68 [DOI] [PubMed] [Google Scholar]
- Jones SM, Klinghoffer R, Prestwich GD, Toker A, Kazlauskas A. PDGF induces an early and a late wave of PI 3-kinase activity, and only the late wave is required for progression through G1. Curr Biol. 1999;9:512-21 [DOI] [PubMed] [Google Scholar]
- Stacey D, Kazlauskas A. Regulation of Ras signaling by the cell cycle. Curr Opin Genet Dev. 2002;12:44-6 [DOI] [PubMed] [Google Scholar]
- Auger KR, Serunian LA, Soltoff SP, Libby P, Cantley LC. PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell. 1989;57:167-75 [DOI] [PubMed] [Google Scholar]
- Whiteford CC, Best C, Kazlauskas A, Ulug ET. D-3 phosphoinositide metabolism in cells treated with platelet-derived growth factor. Biochem J. 1996;319(Pt 3):851-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalaf WF, White H, Wenning MJ, Orazi A, Kapur R, Ingram DA. K-Ras is essential for normal fetal liver erythropoiesis. Blood. 2005;105:3538-41 [DOI] [PubMed] [Google Scholar]
- Zhang J, Lodish HF. Identification of K-ras as the major regulator for cytokine-dependent Akt activation in erythroid progenitors in vivo. Proc Natl Acad Sci U S A. 2005;102:14605-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond TD, Chohan M, Barber DL. Turning cells red: signal transduction mediated by erythropoietin. Trends Cell Biol. 2005;15:146-55 [DOI] [PubMed] [Google Scholar]
- Schmidt EK, Fichelson S, Feller SM. PI3 kinase is important for Ras, MEK and Erk activation of Epo-stimulated human erythroid progenitors. BMC Biol. 2004;2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhu S, Gong Z, Low BC. K-ras/PI3K-Akt signaling is essential for zebrafish hematopoiesis and angiogenesis. PLoS One. 2008;3:e2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandzioch E, Edling CE, Palmer RH, Carlsson L, Hallberg B. Activation of the MAP kinase pathway by c-Kit is PI-3 kinase dependent in hematopoietic progenitor/stem cell lines. Blood. 2004;104:51-7 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Khwaja A, et al. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457-67 [DOI] [PubMed] [Google Scholar]
- Sheng H, Shao J, DuBois RN. Akt/PKB activity is required for Ha-Ras-mediated transformation of intestinal epithelial cells. J Biol Chem. 2001;276:14498-504 [DOI] [PubMed] [Google Scholar]
- Li W, Zhu T, Guan KL. Transformation potential of Ras isoforms correlates with activation of phosphatidylinositol 3-kinase but not ERK. J Biol Chem. 2004;279:37398-406 [DOI] [PubMed] [Google Scholar]
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988-1004 [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424-30 [DOI] [PubMed] [Google Scholar]
- Jia S, Roberts TM, Zhao JJ. Should individual PI3 kinase isoforms be targeted in cancer? Curr Opin Cell Biol. 2009;21:199-208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simi L, Pratesi N, Vignoli M, et al. High-resolution melting analysis for rapid detection of KRAS, BRAF, and PIK3CA gene mutations in colorectal cancer. Am J Clin Pathol. 2008;130:247-53 [DOI] [PubMed] [Google Scholar]
- Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RK, Baker AC, Debiasi RM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347-51 [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Shigematsu H, Nomura M, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008;68:6913-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakoff SJ, Engelman JA, Irie HY, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992-1000 [DOI] [PubMed] [Google Scholar]
- Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102:18443-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenoue T, Kanai F, Hikiba Y, et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;65:4562-7 [DOI] [PubMed] [Google Scholar]
- Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102:802-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels Y, Diaz LA, Jr., Schmidt-Kittler O, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561-73 [DOI] [PubMed] [Google Scholar]
- Bader AG, Kang S, Vogt PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci U S A. 2006;103:1475-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007;104:5569-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008;27:5486-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A. 2008;105:2652-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Vogt PK. Hot-spot mutations in p110alpha of phosphatidylinositol 3-kinase (pI3K): differential interactions with the regulatory subunit p85 and with RAS. Cell Cycle. 2010;9:596-600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111-6 [DOI] [PubMed] [Google Scholar]
- Oda K, Okada J, Timmerman L, et al. PIK3CA cooperates with other phosphatidylinositol 3’-kinase pathway mutations to effect oncogenic transformation. Cancer Res. 2008;68:8127-36 [DOI] [PubMed] [Google Scholar]
- Velho S, Oliveira C, Ferreira A, et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer. 2005;41:1649-54 [DOI] [PubMed] [Google Scholar]
- Rao KN. The significance of the cholesterol biosynthetic pathway in cell growth and carcinogenesis (review). Anticancer Res. 1995;15:309-14 [PubMed] [Google Scholar]
- Tosi MR, Tugnoli V. Cholesteryl esters in malignancy. Clin Chim Acta. 2005;359:27-45 [DOI] [PubMed] [Google Scholar]
- Calleros L, Sanchez-Hernandez I, Baquero P, Toro MJ, Chiloeches A. Oncogenic Ras, but not (V600E)B-RAF, protects from cholesterol depletion-induced apoptosis through the PI3K/AKT pathway in colorectal cancer cells. Carcinogenesis. 2009;30:1670-7 [DOI] [PubMed] [Google Scholar]
- Zhao JJ, Cheng H, Jia S, et al. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci U S A. 2006;103:16296-300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torbett NE, Luna-Moran A, Knight ZA, et al. A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem J. 2008;415:97-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S, Liu Z, Zhang S, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuriato S, Rabin K, Fan AC, Shachaf CM, Felsher DW. Conditional animal models: a strategy to define when oncogenes will be effective targets to treat cancer. Semin Cancer Biol. 2004;14:3-11 [DOI] [PubMed] [Google Scholar]
- Chin L, Tam A, Pomerantz J, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468-72 [DOI] [PubMed] [Google Scholar]
- Lim KH, Counter CM. Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell. 2005;8:381-92 [DOI] [PubMed] [Google Scholar]
- Lim KH, Ancrile BB, Kashatus DF, Counter CM. Tumour maintenance is mediated by eNOS. Nature. 2008;452:646-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher GH, Wellen SL, Klimstra D, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan A, Weinberg RA. Opinion. Comparative biology of mouse versus human cells: modelling human cancer in mice. Nat Rev Cancer. 2003;3:952-9 [DOI] [PubMed] [Google Scholar]
- Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. 2004;6:171-83 [DOI] [PubMed] [Google Scholar]
- Hamad NM, Elconin JH, Karnoub AE, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002;16:2045-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895-904 [DOI] [PubMed] [Google Scholar]
- Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563-72 [DOI] [PubMed] [Google Scholar]
- Duffy MJ, McGowan PM, Gallagher WM. Cancer invasion and metastasis: changing views. J Pathol. 2008;214:283-93 [DOI] [PubMed] [Google Scholar]
- Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274-84 [DOI] [PubMed] [Google Scholar]
- Giehl K. Oncogenic Ras in tumour progression and metastasis. Biol Chem. 2005;386:193-205 [DOI] [PubMed] [Google Scholar]
- Campbell PM, Der CJ. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin Cancer Biol. 2004;14:105-14 [DOI] [PubMed] [Google Scholar]
- Sasaki AT, Firtel RA. Regulation of chemotaxis by the orchestrated activation of Ras, PI3K, and TOR. Eur J Cell Biol. 2006;85:873-95 [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629-35 [DOI] [PubMed] [Google Scholar]
- Jiang K, Sun J, Cheng J, Djeu JY, Wei S, Sebti S. Akt mediates Ras downregulation of RhoB, a suppressor of transformation, invasion, and metastasis. Mol Cell Biol. 2004;24:5565-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Hu D, Tu L, et al. Involvement of PI3K/Akt signaling pathway in hepatocyte growth factor-induced migration of uveal melanoma cells. Invest Ophthalmol Vis Sci. 2008;49:497-504 [DOI] [PubMed] [Google Scholar]
- Shaw LM, Rabinovitz I, Wang HH, Toker A, Mercurio AM. Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell. 1997;91:949-60 [DOI] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599-602 [DOI] [PubMed] [Google Scholar]
- Kim D, Kim S, Koh H, et al. Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase production. FASEB J. 2001;15:1953-62 [DOI] [PubMed] [Google Scholar]
- Park BK, Zeng X, Glazer RI. Akt1 induces extracellular matrix invasion and matrix metalloproteinase-2 activity in mouse mammary epithelial cells. Cancer Res. 2001;61:7647-53 [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409-16 [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56-68 [DOI] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539-44 [DOI] [PubMed] [Google Scholar]
- Schober M, Raghavan S, Nikolova M, et al. Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J Cell Biol. 2007;176:667-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watermann DO, Gabriel B, Jager M, et al. Specific induction of pp125 focal adhesion kinase in human breast cancer. Br J Cancer. 2005;93:694-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan R, Smolkin MB, Cocker R, Fayyad R, Oktay MH. Focal adhesion proteins as markers of malignant transformation and prognostic indicators in breast carcinoma. Hum Pathol. 2006;37:9-15 [DOI] [PubMed] [Google Scholar]
- Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest. 2009;119:252-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi P, Giannoni E. Anoikis: a necessary death program for anchorage-dependent cells. Biochem Pharmacol. 2008;76:1352-64 [DOI] [PubMed] [Google Scholar]
- Zhan M, Zhao H, Han ZC. Signalling mechanisms of anoikis. Histol Histopathol. 2004;19:973-83 [DOI] [PubMed] [Google Scholar]
- Simpson CD, Anyiwe K, Schimmer AD. Anoikis resistance and tumor metastasis. Cancer Lett. 2008;272:177-85 [DOI] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 1997;16:2783-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang F, Lee JT, Navolanic PM, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590-603 [DOI] [PubMed] [Google Scholar]
- Rommel C, Clarke BA, Zimmermann S, et al. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286:1738-41 [DOI] [PubMed] [Google Scholar]
- Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science. 1999;286:1741-4 [DOI] [PubMed] [Google Scholar]
- Menges CW, McCance DJ. Constitutive activation of the Raf-MAPK pathway causes negative feedback inhibition of Ras-PI3K-AKT and cellular arrest through the EphA2 receptor. Oncogene. 2008;27:2934-40 [DOI] [PubMed] [Google Scholar]
- Reusch HP, Zimmermann S, Schaefer M, Paul M, Moelling K. Regulation of Raf by Akt controls growth and differentiation in vascular smooth muscle cells. J Biol Chem. 2001;276:33630-7 [DOI] [PubMed] [Google Scholar]
- Laprise P, Langlois MJ, Boucher MJ, Jobin C, Rivard N. Down-regulation of MEK/ERK signaling by E-cadherin-dependent PI3K/Akt pathway in differentiating intestinal epithelial cells. J Cell Physiol. 2004;199:32-9 [DOI] [PubMed] [Google Scholar]
- Schulze A, Lehmann K, Jefferies HB, McMahon M, Downward J. Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev. 2001;15:981-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballif BA, Roux PP, Gerber SA, MacKeigan JP, Blenis J, Gygi SP. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proc Natl Acad Sci U S A. 2005;102:667-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648-57 [DOI] [PubMed] [Google Scholar]
- Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179-93 [DOI] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151-62 [DOI] [PubMed] [Google Scholar]
- Zhang H, Bajraszewski N, Wu E, et al. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650-6 [DOI] [PubMed] [Google Scholar]
- O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PM, Groehler AL, Lee KM, Ouellette MM, Khazak V, Der CJ. K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res. 2007;67:2098-106 [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11-22 [DOI] [PubMed] [Google Scholar]
- Garcia-Echeverria C. Protein and lipid kinase inhibitors as targeted anticancer agents of the Ras/Raf/MEK and PI3K/PKB pathways. Purinergic Signal. 2009;5:117-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramjaun AR, Downward J. Ras and phosphoinositide 3-kinase: partners in development and tumorigenesis. Cell Cycle. 2007;6:2902-5 [DOI] [PubMed] [Google Scholar]
- Halilovic E, She QB, Ye Q, et al. PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res. 2010;70:6804-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee S, Jagani Z, Xiang KX, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286-93 [DOI] [PubMed] [Google Scholar]
- Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkade CW, Castillo-Martin M, Puzio-Kuter A, et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008;118:3051-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KA, Yeager N, Baker K, Liao XH, Refetoff S, Di Cristofano A. Oncogenic Kras requires simultaneous PI3K signaling to induce ERK activation and transform thyroid epithelial cells in vivo. Cancer Res. 2009;69:3689-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton JG, Steelman LS, Lee JT, et al. Effects of the RAF/MEK/ERK and PI3K/AKT signal transduction pathways on the abrogation of cytokine-dependence and prevention of apoptosis in hematopoietic cells. Oncogene. 2003;22:2478-92 [DOI] [PubMed] [Google Scholar]